










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.38 n.1 Pretoria Jan. 2012
Application of magnesium hydroxide and barium hydroxide for the removal of metals and sulphate from mine water
V Bologo*; JP Maree; F Carlsson
Department Environmental, Water and Earth Sciences, Tshwane University of Technology, Private Bag X680, Pretoria 0001, South Africa
ABSTRACT
The proposed magnesium-barium-oxide process consists of metal removal with Mg(OH)2, magnesium and sulphate removal with Ba(OH)2 and calcium removal with CO2. The raw materials, Mg(OH)2 and Ba(OH)2 are recovered from the BaSO4 and Mg(OH)2 sludges that are produced. Laboratory studies showed that metals are removed to low levels. This includes iron(II), the dominant metal ion in mine water, which is first oxidised to iron(III), whereafter it precipitates as Fe(OH)3 resulting in residual levels of Fe(II) in the mine water of less than 20 mg/ℓ. Sulphate is also removed to less than 25 mg/ℓ. The final sulphate concentration is a function of the amount of Ba(OH)2 dosed, as the amount of sulphate removed is stoichiometrically equivalent to the Ba(OH)2 dosage. During CO2-dosing, CaCO3 is precipitated to the saturation level of CaCO3.
Keywords: Magnesium hydroxide; barium hydroxide; sulphate removal; water treatment
Introduction
Mining is a significant contributor to water pollution owing primarily to pyrite oxidation that generates potentially high levels of acidity, metal ions (mainly Fe), and sulphate (Reaction (1)) (Barbes and Romberger, 1968):
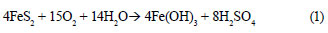
South Africa';s Witwatersrand Basin produces 340 Mℓ/d of mine-water, of which 50 Mℓ/d is produced by the Western Basin. Most of the underground mining operations in the Western Basin were closed by 1990. By 2004 the underground voids were filled by rising water levels and acid water started to decant at the surface. Contaminated void spaces increase with time and result in: (a) less water suitable for irrigation of crops, (b) less potable water available for the growing local communities, (c) the natural drying up of rivers, and (d) stream bed losses. The water of mining origin in the Western Basin contains, on average, 4 800 mg/ℓ sulphate, 800 mg/ℓ Fe(II), 100 mg/ℓ Fe(III), 230 mg/ℓ Mn, 11 mg/ℓ Zn, 18 mg/ℓ Ni, 5 mg/ℓ Co, 6 mg/ℓ Al, 150 mg/ℓ Mg, some radioactivity and 700 mg/ℓ free acidity (as CaCO3). This acid mine drainage (AMD) source creates a major concern in that the Cradle of Humankind, a World Heritage Site, is not far from the decant point. As of March 2010, AMD started decanting into the Krugersdorp Nature Reserve at a rate of between 10 and 60 Mℓ/day, with the possibility of reaching the Sterkfontein Cave System which includes the Cradle of Humankind, where the earliest known hominid fossil remains were discovered and where paleontological excavations continue (Zvinowanda et al., 2010).
It is essential that a technically sound and cost-effective solution be found for the acid mine-water problem. South Africa is water-constrained and also has a mature mining economy with numerous mines having closed or in the process of being decommissioned. Should a solution be found that generates income, the AMD problem could be solved via economic principles rather than through government intervention by means of policies and legislated control measures.
Several processes have been considered for sulphate removal, e.g. biological removal, SAVMIN (etringite) (Ramsay, 1998), ecoDose, reverse osmosis (Chamber of Mines Research Organisation, 1988), and electrodialysis (Pulles et al., 1996). Soluble barium salts can also be used for sulphate removal and have certain advantages in that sulphate can be removed to specific levels owing to the low solubility of barium sulphate (Volman, 1984; Maree et al., 1989; Adlem et al., 1991). Soluble barium salts, such as barium sulphide, barium chloride and barium hydroxide, are potential candidates for the treatment regime and can be regenerated at the end of the process.
Kun (1972) studied the removal of sulphate with barium carbonate (BaCO3). He identified 3 problems: a long retention time is required; high concentrations of soluble barium remain in the treated water when excess BaCO3 is dosed over the required stoichiometric amount; and, the high cost of BaCO3. Volman (1984) and Maree et al. (1989) overcame the cost problem by demonstrating that barium sulphate (BaSO4) could be reduced efficiently and economically with coal under thermal conditions to produce barium sulphide (BaS). This compound can be used directly for the process or first converted to BaCO3. Wilsenach (1986) demonstrated the economic viability of the process by calculating the cost of producing BaS from BaSO4.
Trusler et al. (1988) developed an integrated process which includes the recovery of BaCO3 and lime. They noted that incomplete sulphate removal with BaCO3 resulted when sulphate is not completely associated with calcium ions. They overcame this problem by dosing lime for pre-removal of magnesium as Mg(OH)2. Maree et al. (1989) have developed a barium carbon ate process (a 2-stage, fluidised-bed reactor system) to overcome the other problems identified by Kun (1972), i.e., the long retention time and the high barium concentration in the treated water. A disadvantage of the barium carbonate process, proposed by Trusler (1988) and improved by Maree et al. (1989), is the formation of a mixed precipitate of BaSO4 and CaCO3. Bosman et al. (1990) and Adlem et al. (1991) overcame this problem by pre-treating the AMD with lime to remove metals before the addition of barium sulphide. BaS is highly soluble in water and can therefore readily provide Ba2+ ions for fast interaction with SO42- in sulphate-rich water. Maree et al. (1990) elucidated the ability of BaS to directly remove sulphate from acid waters. Other studies have demonstrated that BaS has an advantage of producing by-products like sulphur and NaHS, which can potentially be derived from the H2S produced during the H2S stripping stage, and CaCO3 from the softening stage (Maree et al., 1989; Bosman et al., 1990; Adlem et al., 1991; Du Preez and Maree, 1994). Laboratory studies were carried out by Maree et al. (2004) to demonstrate that the integrated BaS process is technically and economically viable for sulphate removal. This process consists of 4 stages. In the thermal stage, BaSO4 is reduced to BaS at 1 050ºC, in a kiln using coal as the reductant. In the sulphate removal stage, sulphate is precipitated as BaSO4. In the stripping stage, the barium sulphide is dissolved in water and reacted with CO2 from the kiln to remove or strip off the H2S gas. In the softening stage, CaCO3 is precipitated as a result of CO2-stripping with air.
Hlabela et al. (2007) demonstrated that BaCO3 can be used effectively for sulphate removal after pre-treatment of the AMD with lime for removal of metals, including magnesium, as metal hydroxides.
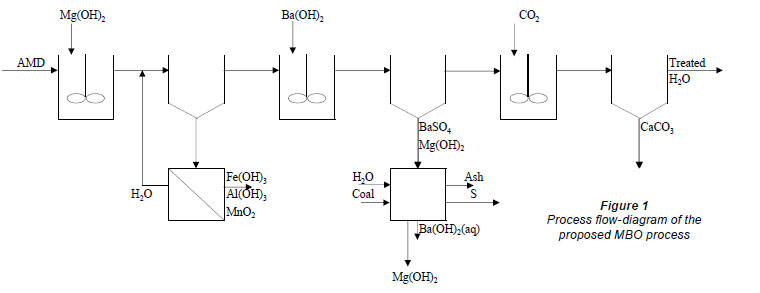
The purpose of the present study was to demonstrate, using a laboratory study, the performance of an alternative, integrated magnesium hydroxide and barium hydroxide process (MBO Process) for treatment of acid mine-water. The provisionally patented process (Maree and Louw, 2010) consists of the following stages (Fig. 1):
• Magnesium hydroxide, for neutralisation and metal removal
• Barium hydroxide, for sulphate and magnesium removal
• CO2, for CaCO3 precipitation
• Recovery of Ba(OH)2 via BaS from the BaSO4 produced
The specific aims were to demonstrate that:
• Metals can be completely removed with Mg(OH)2
• Sulphate concentration can be lowered to less than 200 mg/ℓ
• Magnesium concentration can be lowered to less than 10 mg/ℓ with Ba(OH)2
• The high pH water can be stabilised with CO2
The processing of BaSO4 and Mg(OH)2 in the sludge to recover the process raw materials, Ba(OH)2 and Mg(OH)2, were investigated separately.
Materials and methods
Feedstock
Waters from a coal mine and a gold mine were used as feed waters containing 2 400 - 4 800 mg/ℓ sulphate (for chemical analyses, see Tables 1 and 2). Magnesium hydroxide and barium hydroxide were used for pH adjustment and sulphate removal, respectively. CO2 gas (Afrox) was used for pH adjustment after sulphate removal.
The following chemicals were used in this study: Barium hydroxide (Ba(OH)2.8H2O, analytical reagent, Merck) and calcium hydroxide (97% Ca(OH)2, Rochelle Chemicals, Johannesburg); Mg(OH)2 (CP grade, May and Baker Ltd, Dagenham, England), air (HP compressed; Afrox, South Africa), and CO2 (Afrox, South Africa).
Equipment
Neutralisation with Mg(OH)2 and sulphate removal with Ba(OH)2 were studied using stirred beakers.
Experimental procedure
Feed water was mixed in a beaker (5 ℓ), aerated for iron(II) oxidation and stirred (30 min) for completion of the oxidation/precipitation reactions. The sludge was allowed to settle and the clear water decanted. Ba(OH)2 was mixed with the decant water (1000 mℓ glass beakers). Stabilisation of the Ba(OH)2 treated water was achieved by passing CO2 through the water.
Experimental programme
The following parameters were investigated: metal removal with Mg(OH)2, magnesium and sulphate removal with Ba(OH)2, and calcium removal with CO2.
Analytical
Samples were collected regularly and filtered (Whatman No. 1). Sulphate, alkalinity, Fe(II), mixed liquor suspended solids (MSS), volatile suspended solids (VSS), acidity, and pH were determined according to standard procedures (APHA, 1989). Calcium and magnesium were assayed using atomic absorption spectrophotometry.
Results and discussion
Water quality and chemical reactions
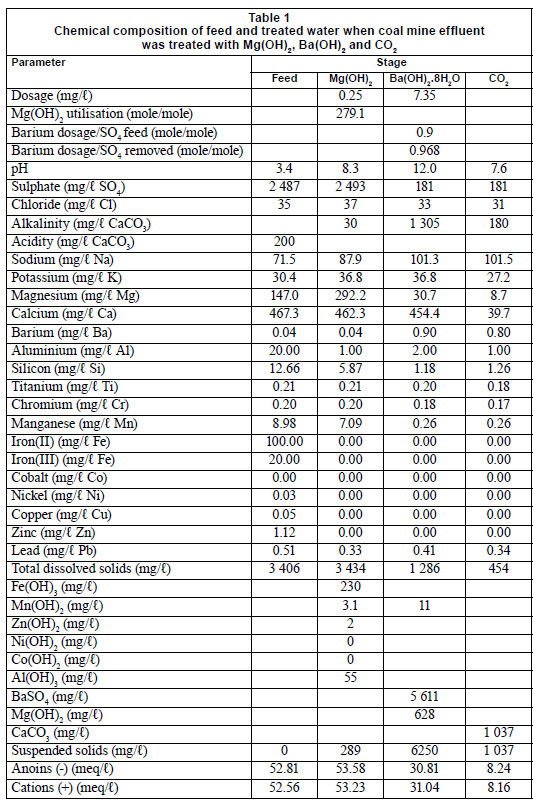
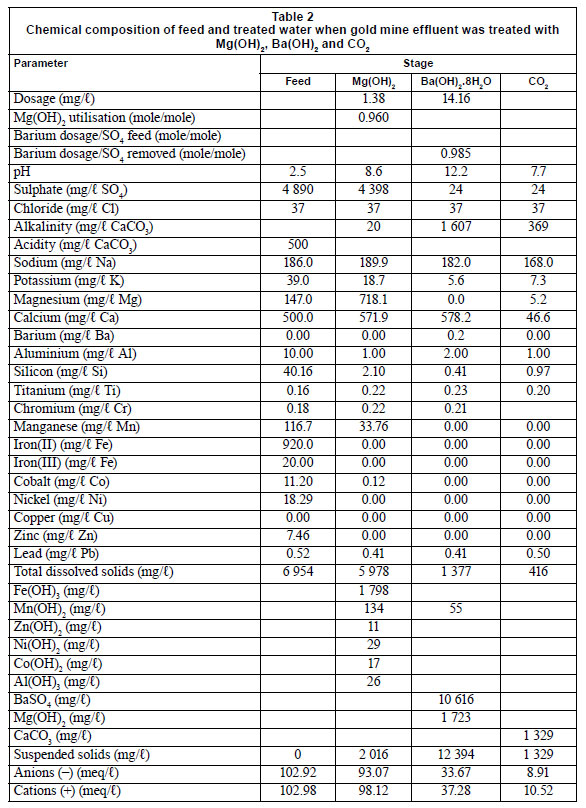
The MBO study can be applied to the treatment of acid mine-water. Tables 1 and 2 show the chemical composition of the feed waters before and after treatment with Mg(OH)2, Ba(OH)2 and CO2, for effluents from coal and gold mines, respectively. The total dissolved solids (TDS) content in the case of coal mine-water was 3 406 mg/ℓ in the feed water, 3 434 mg/ℓ after Mg(OH)2 treatment, 1 286 mg/ℓ after Ba(OH)2 treatment and 454 mg/ℓ after CO2 addition (Table 1). Similarly, in the case of gold mine effluent, the overall TDS content was lowered from 6 954 mg/ℓ in the feed water to 416 mg/ℓ, following treatment with the MBO process (Table 2). During Mg(OH)2 treatment, free acid and all of the metal concentrations, excluding that of calcium and magnesium, were lowered to below allowable limits for drinking water in South Africa (SANS 241:2006). The lowering of the TDS in the case of Mg(OH)2 treatment was mainly due to the oxidation of Fe2+ to Fe3+ (Reaction (2)) and precipitation as Fe(OH)3 (Reaction (3)). Similarly, Mn2+ was oxidised to Mn4+ and precipitated as MnO2. The other metals, Al3+, Co2+, Zn2+, Pb2+, Co2+ and Ni2+, precipitated as metal hydroxides. This was owing to the low solubility-products for Fe(OH)(2.64 x 10-39); Al(OH)(8.5 x 10-23); Mn(OH)(2.06 x 10-13); Cu(OH)2 (2.20 x 10-20); Zn(OH)2 (7.71 x 10-17); Pb(OH)2 (1.42 x 10-20); Co(OH)2 (1.09 x 10-15) and Ni(OH)2 (5.47 x 10-16) (Sillen and Martell, 1964; Lide, 1992).
The precipitated BaSO4 and Mg(OH)2 can be made to settle rapidly by dosing coagulants. Coagulant dosing can be avoided by controlling the precipitation process to yield particles with improved settling rates. Hlabela (2009) showed that a multiple point, in-line dosing system can produce flocs within the size range 10 - 100 µm. In the final step, the calcium concentration in the feed water was lowered from 454 mg/ℓ down to 40 mg/ℓ (as Ca+) by stabilising the water with CO2 (Reaction (8)). During CO2 addition for pH adjustment, CaCO3 crystallisation occurred, as indicated by the decrease in the calcium concentration. According to Loewenthal et al. (1986), the saturation, under-saturation and super-saturation states of CaCO3 are theoretically identified by the activity product of Ca2+ and CO32- that is comparable to the solubility product constant, ksp, for CaCO3. Above the solubility product, CaCO3 will precipitate from solution, and below, CaCO3 will dissolve. The rate of precipitation or dissolution is described by Eq. (9), where k is the precipitation rate-constant, S, the surface area of CaCO3 growth/dissolution sites, and the parameters in square brackets, activities in mol/ℓ. The rate of precipitation depends on k, S and the term within curly brackets. Although k is affected to an unknown extent by crystal structure, S is dependent on the size, mass and structure of the crystals. By controlling S and the degree of super-saturation (term in curly brackets), the rate of precipitation or dissolution of CaCO3 can be determined (Loewenthal et al., 1986).
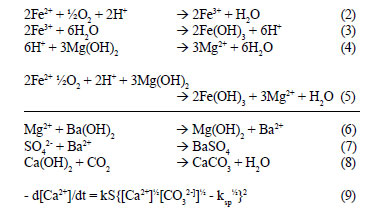
where:
k = precipitation rate-constant,
S = surface area of CaCO3 growth/dissolution sites and the terms in square brackets, activities in mol/ℓ.
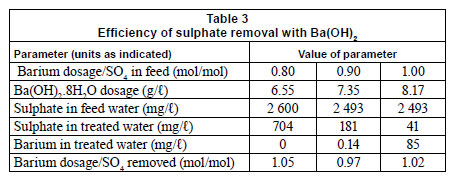
The rate of sulphate removal through BaSO4 crystallisation is influenced by the sulphate concentration in solution and the BaSO4 seed crystal concentration. It was observed that sulphate removal was stoichiometrically equivalent to the Ba(OH)2 dosage, as indicated by [Ba2+ dosage]/[SO4 removed] (mol/mol) ratios of 1.05, 0.97 and 1.02, respectively (Table 3). As expected, higher residual Ba2+ concentrations were in solution with lower residual SO42 concentrations. This is due to the low solubility product of BaSO4 and the rate of BaSO4 crystallisation at low seed-crystal concentrations. The practical implication is that about 200 mg/ℓ free sulphate should remain in solution to limit Ba2+ concentration to less than 2 mg/ℓ in solution, the guideline value for drinking water as laid down by the USEPA (2011). Optimum conditions also need to prevail to allow rapid BaSO4-crystallisation, such as providing a solid BaSO4 presence in the reactor by means of sludge recirculation.
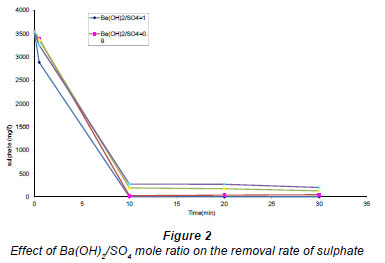
Barium ions react with sulphate ions forming a white precipitate of BaSO. Figure 2 shows an increase in the sulphate removal rate with increased barium hydroxide concentration (molar ratios of 0.8, 0.85, 0.9 and 1) in the solution. From chemical Reaction (7), 1 mole of barium reacts with 1 mole of sulphate. Therefore, it is clear that the more barium ions available in solution the more sulphate ions are removed from the solution. Sulphate removal followed normal stoichiometry. Therefore, it is sufficient to use a 1:1 mole ratio of dissolved barium to sulphate for almost complete removal of sulphate from wastewater.
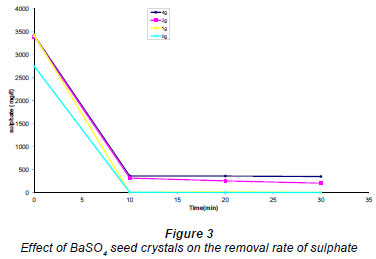
The effect of BaSO4 seeds added to AMD before the addition of Ba(OH)2 was investigated. The results are shown in Fig. 3. From these results it was observed that the presence of a small amount of barium sulphate seed did not have any significant effects on the removal of sulphate.
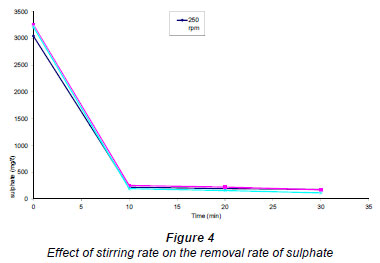
Four different experiments were carried out using increasingly rapid stirring rates. The results are shown in Fig. 4. It was also observed (Fig. 4) that the removal-rate initially increased linearly and subsequently flattened to become abruptly constant, showing a complete removal of sulphate. The stirring rates in the range 250-800 r/min did not show any significant differences in the removal rates of sulphates.
An advantage of using Mg(OH)2 for neutralisation and metal removal is that the precipitated metal hydroxides are not mixed with gypsum. A sludge of higher density was produced which settles to the bottom faster. The sludge from the Mg(OH)2 stage consisted of Fe(OH)3, Al(OH)3, MnO2 and Zn(OH)2. The 'Ba(OH)2 sludge'; consisted of 70% BaSO4 and 30% Mg(OH)2.
Conclusions
With the MBO study at a laboratory scale, it was demonstrated, that:
• Mg(OH)2 precipitated metals as metal hydroxides. Iron(II) was first oxidised to iron(III) before being precipitated.
• Ba(OH)2 precipitated sulphate as BaSO4 and magnesium as Mg(OH)2. Sulphate removal was stoichiometrically equivalent to the Ba(OH)2 dosage.
• 2 precipitated calcium as CaCO3.
Acknowledgements
This work was made possible with the support of the following institutions: The National Research Foundation, which provided funding for Tshwane University of Technology projects on neutralisation and sulphate removal through their Technology and Human Resources for Industry Programme (THRIP); Rand Water, which funds activities falling under the Rand Water Chair in Water Utilisation at the Tshwane University of Technology, and Tshwane University of Technology, which provided substantial financial support for the research programme. The contributions of the following people are gratefully acknowledged: Dr Fritz Carlsson for editing the manuscript and Dr Caliphs Zvinowanda, Mr Kebede Kefeni and Mr Dave Katlekgo Mpholoane for assistance with the laboratory work.
References
ADLEM CJ, MAREE JP and DU PLESSIS P (1991) Treatment of sulphate-rich mining effluents with the barium hydroxide process and recovery of valuable by-products. Proc. 4th International Mine Water Assoc. Congress, 25-30 September 1991, Ljubljana (Slovenia)-Portschach (Austria). 211-222. [ Links ]
APHA (1989) Standard Methods for the Examination of Water and Wastewater (17th edn.). American Pubic Heath Association, New York. [ Links ]
BARBES HJ and ROMBERGER SB (1968) Chemical aspects of acid mine drainage. J. Water Poll. Contr. Fed. 40 (3) 371-384. [ Links ]
BOSMAN DJ, CLAYTON JA, MAREE JP and ADEM CJ (1990). Removal of sulphate from mine water with barium sulphide. Mine Water Env. 9 (1-4) 149-163. [ Links ]
CHAMBER OF MINES RESEARCH ORGANISATION (1988) New desalination program on stream. R&D News CM, October 1988. [ Links ]
DU PREEZ A and MAREE JP (1994) Pilot-scale biological sulphate and nitrate removal utilizing producer gas as energy source. Proc. 7th International Symposium on Anaerobic Digestion, Cape Town, South Africa, 23-28 January. 190-204. [ Links ]
HLABELA PS, MAREE JP and BRUINSMA D (2007) Barium carbonate process for sulphate and metal removal from mine water. Mine Water Environ. 26 (1) 14-22. [ Links ]
HLABELA PS (2009) The integrated barium carbonate process for sulphate removal from acid mine water. Ph.D. thesis (Chemical Engineering), North-West University (Potchefstroom Campus). 152 pp. [ Links ]
KUN E (1972) A report on the reduction of the sulphate content of acid mine drainage by precipitation with barium carbonate. Internal Report, Anglo American Research Laboratories, Project D/3/W/1. [ Links ]
LIDE DR (1992) Handbook of Chemistry and Physics (73rd edn.). CRC Press, Boca Raton. [ Links ]
LOEWENTHAL RE, WIECHERS HNS, MARAIS GvR (1986) Softening and Stabilization of Municipal Waters. WRC Report No. TT 24/86. Water Research Commission, Pretoria. [ Links ]
MAREE JP (1989) Treatment of water (BaS process), Republic of South Africa (Patent No. 90/1889), 28 November ), Canada (Patent No: 2 012 266 8) and United States of America (Patent No. 5,035,807). [ Links ]
MAREE JP, BOSMAN DJ and JENKINS GR (1989) Chemical removal of sulphate, calcium and heavy metals from mining and power station effluents. Proc. 1st Biennial Conf. of the Water Inst. Of Southern Africa, 29 to 30 March 1989, Cape Town. [ Links ]
MAREE JP, LEIBOWITZ A and DODS D (1990) Sulphate wastes. Symposium on Removal of Sulphate from Mine Water, Water Inst. Of Southern Africa, 8 June 1990, Rustenburg Platinum Mines and Base Mineral Refinery Club, Rustenburg. [ Links ]
MAREE JP, HLABELA P, NENGOVHELA R, GELDENHUYS AJ, MBHELE N, NEVHULAUDZI T and WAANDERS FB (2004) Treatment of mine water for sulphate and meta removal using barium sulphide. Mine Water Env. 23 (4) 195-203. [ Links ]
MAREE JP and LOUW WF (2010) Treatment of water (magnesiumbarium-oxide process). Provisional patent application 2011/02173 (25 March 2010), Republic of South Africa. [ Links ]
PULLES W, HOWIE D, OTTO D and EASTON J (1996) A Manual on Mine Water Treatment and Management Practices in South Africa. Appendix Vol. 1: Literature reviews. WRC Report No. 527/1/96. Water Research Commission, Pretoria. [ Links ]
RAMSAY IJ (1998) Process for the treatment of water streams. South African patent application, Number 98/4724. [ Links ]
SOUTH AFRICAN NATIONAL STANDARD (SANS) 241:2006: Drinking water, Edition 6.1. SABS Standards Division, Pretoria. [ Links ]
SILLEN G and MARTELL AE (1964) Stability constants of metal ligand complexes, Spec. Pub. 17. The Chemical Society, London. [ Links ]
TRUSLER GE, EDWARDS RI, BROUCKAERT CJ and BUCKLEY CA (1988) The chemical removal of sulphates. Proc. 5th National Meeting of the S.A. Institute of Chemical Engineers, 15-16 August 1988, Pretoria. W3-0-W3-11. [ Links ]
USEPA (2011) Water: Drinking Water Contaminants. National Primary Drinking Water Regulations. URL: http://water.epa.gov/drink/contaminants/index.cfm (Accessed 11-01-2011). [ Links ]
VOLMAN R (1984) The use of barium sulphide to remove sulphate from industrial effluents. M.Sc. Thesis (Chem. Eng.), University of Stellenbosch. [ Links ]
WEAST RC (1971) Handbook of Chemistry and Physics (52nd edn.). The Chemical Rubber Co., Boca Raton. p. B107. [ Links ]
WILSENACH JT (1986) Cost estimate for barium sulphate reduction. Internal report, Div. of Water Technology, CSIR, Project No. 620/2616/6. [ Links ]
ZVINOWANDA CM, MAREE JM and VAN DER WALT B (2010) Temporary neutralization plant. Rand Uranium, April 2010. Progress Report No 1. [ Links ]
Received 16 February 2011; accepted in revised form 14 December 2011.
* To whom all correspondence should be addressed. +27 12 382 6320; fax: +27 12 382-6354; e-mail: bologov@tut.ac.za


{kind=link}
{kind=link}