










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.37 n.3 Pretoria Jul. 2011
Insights into the bacterial diversity in a freshwater-deprived permanently open Eastern Cape estuary, using 16S rRNA pyrosequencing analysis
GF MatcherI,*; RA DorringtonI; TO HenningerII; PW FronemanII
IDepartment of Biochemistry, Microbiology and Biotechnology, Rhodes University, PO Box 94, Grahamstown, 6140, South Africa
IIDepartment of Zoology and Entomology, Rhodes University, PO Box 94, Grahamstown, 6140, South Africa
ABSTRACT
The aim of this study was to conduct an investigation into the bacterial diversity in the freshwater-deprived Kariega Estuary, situated along the Eastern Cape coastline, using ribosomal RNA gene sequences obtained by pyrosequencing. Shifts in the microbial diversity were correlated to selected physico-chemical variables along the length of the estuary. More than 27 000 sequences were obtained and rarefaction analyses confirmed a comprehensive appraisal of the microbial diversity present in the Kariega Estuary. Distinct patterns in phylotype distribution from the hypersaline upper reaches to the mouth of the estuary were observed; notably, the importance of the detrital food web within the Kariega Estuary was highlighted by the high occurrence of Bacteroidetes and Actinomycetes. Moreover, while the observed chlorophyll-a concentrations were low (< 0.1 µg·ℓ-1), the presence of Pelagibacter and Flavobacteria amongst the microbial community suggests a potentially important contribution of these microbes towards the total primary productivity of the ecosystem. No human pathogenic microbes were detected within waters of the system. We conclude that pyrosequencing provides a versatile and efficient tool for assessing the microbial diversity in the Kariega Estuary and propose that this technology may provide valuable information on the ecosystem functioning and health of aquatic ecosystems.
Keywords: permanently open, oligotrophic, estuary, 16S rRNA, pyrosequencing, bacterial biodiversity
Introduction
Prokaryotes are ubiquitous, extremely abundant and diverse, and have been demonstrated to play a major role in regulating key biogeochemical processes, including the carbon and nitrogen cycles in aquatic environments (Kirchman, 2008). Furthermore, it has been recognised that there is a close relationship between the primary productivity of aquatic ecosystems and the species diversity and speciation of numerically dominant families of microbes (Horner-Devine et al., 2003; Smith, 2007). However, analyses of aquatic ecosystems seldom consider microorganisms and their metabolic activity, with little known of the complex factors influencing bacterial community composition or the effect these communities have on ecosystem functioning (Dolan, 2005; Teira et al., 2008). Phylogenetic identification of microorganisms within given environments and correlation of their observed diversity and distribution with the environmental factors in play has thus become a burgeoning and critical field of study. Additionally, human activities in the form of household and industrial wastes and nutrient status alterations impact heavily on aquatic ecosystems. Due to their small size and rapid proliferation rates, microbes respond readily to anthropogenic pollution exhibiting changes in microbial diversity and dominant phylotype representation (Lemke et al., 1997; Ford, 2000).
Culture-independent molecular surveys for characterising microbial communities have become the norm due to the fact that more than 90% of the microbes present in the environment are difficult to culture or are unculturable (Schloss and Handelsman, 2005). The mainstay of the molecular techniques for microbial identification is sequence analysis of the 16S ribosomal RNA (16S rRNA) gene. The 16S rRNA gene is tailor-made for microbial identification due to its universal presence in bacteria, extreme species sequence conservation and evolution-induced interspecies variability (Tringe and Hugenholtz, 2008). The 16S rRNA gene consists of highly-conserved regions interspersed with 9 species-dependent hypervariable regions (V1-V9). While sequencing of the entire 16S rRNA gene remains the gold standard for accurately classifying bacteria, several studies have validated the use of 16S rRNA hypervariable regions for microbial taxonomic analysis of environmental samples (Wang et al., 2007; Liu et al., 2007; Huse et al., 2008; Wang and Qian, 2009; Claesson et al., 2009).
Until very recently, a typical 16S rRNA survey would include traditional Sanger sequencing by capillary electrophoresis of a few hundred cloned sequences, and consequently would only be able to sample the dominant phylotypes within the target population. By contrast, the newly developed pyrosequencing (high throughput) technologies have not only dramatically reduced the cost and time of sequencing but also generate several thousand sequences per sample (Huse et al., 2008). In addition to the identification of dominant bacterial phylogenetic profiles within a given environment, the high volume of sequences produced with pyrosequencing also allows for the detection of the 'rare' biosphere within the bacterial community. This often overlooked 'rare' biosphere may contribute to critical components of complex consortia or, alternatively, be remnants of a historical ecological change with the potential to become dominant in response to shifts in environmental conditions. An additional advantage of pyrosequencing over the limited traditional clone sequencing is that, due to the large number of 16S rRNA genes analysed, a more accurate representation of the relative abundances of the bacterial phylotypes present in a target environment can be generated (Sogin et al., 2006; Huse et al., 2008).
The aim of this study was to conduct a preliminary investigation into the bacterial diversity in the freshwater-deprived Kariega Estuary, situated along the Eastern Cape coastline, using ribosomal RNA gene sequences obtained by pyrosequencing. Shifts in the microbial diversity were correlated to selected physico-chemical variables along the length of the estuary.
Materials and methods
Study site
The permanently open Kariega Estuary (33º 40' 32"S; 26º 40' 34" E), situated approximately 120 km east of Port Elizabeth, lies on the south-eastern coastline of South Africa (Fig. 1). The estuary can be regarded as a homogenous oligotrophic marine system due to the low freshwater input resulting from sporadic rainfall, a small catchment area (≈ 680 km2), as well as several impoundments (3 major dams and numerous small farm weirs) along the Kariega River (Hodgson, 1987; Grange and Allanson, 1995; Froneman, 2000). The freshwater inflow has been estimated at 2.5-35 x 106 m3 per year (Jennings, 2006). The lower reaches of the estuary are characterised by sand flat-borders and salt marshes comprising the high marsh plants Sarcocornia perennis, Chenolea diffusa and Spartina maritima (Hodgson, 1987). Water depth varies from 1.10 (in the upper reaches) to 3.96 m (near the mouth), depending on the tidal state.
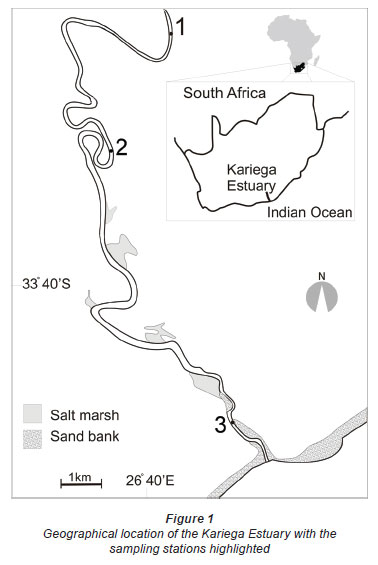
Water samples for the determination of physico-chemical and biological variables and bacterial diversity were collected from 3 sites along the length of the estuary, corresponding to the upper, middle and lower reaches of the system (Fig. 1). Sampling of all 3 sites was done on the same morning.
Physico-chemical variables
Physico-chemical variables were measured at each of the sites in the surface, mid-water and bottom waters. Salinity (parts per thousand (0/00)), water temperature (ºC), dissolved oxygen (mg·ℓ-1) and turbidity values (attenuation coefficients) were measured during high tide employing an Aquaread Aquameter 200 / Aquaprobe 800.
Seston and chlorophyll-a concentrations
Water samples for the determination of particulate organic matter (POM) and total chlorophyll-a (chl-a) concentration were collected from the surface and subsurface (≈ 0.5 m) waters using an 8 ℓ Niskin bottle. Filtration of 500 mℓ water samples through pre-weighed GF/F filters (Sartorius) allowed the determination of the seston concentration (total particulates in suspension) at the different depths and stations. Total chl-a concentrations were determined by filtering a 200 mℓ water sample from each depth and station through GF/F filters and extracting the chlorophyll in 90% acetone in the dark at -20ºC for 24 h. A Turner 10AU fluorometer was then employed to determine the total chl-a concentration before and after acidification, according to the method of Holm-Hansen and Riemann (1978). Chl-a concentrations were expressed as µg chl-a·ℓ-1 and POM concentrations as mg·ℓ-1.
Genomic DNA extraction
Total genomic DNA was isolated, in duplicate, from the 3 sites. Surface water samples (200 mℓ) were initially filtered through a 1 mm mesh to remove large debris, before passing through a 0.22 µm filter. The 0.22 µm filter was then processed using the PowerWater DNA Isolation Kit (MoBio Laboratories) yielding purified genomic DNA for use in PCR amplification.
PCR template preparation and multiplex pyrosequencing
The primer pair E517F (5'-CAGCAGCCGCGGTAA-3') and E969-984 (5'-GTAAGGTTCYTCGCGT-3') was selected for amplification of variable regions 4 and 5 of the bacterial 16S rRNA gene, as it has been shown to have high coverage in all known phyla (Wang et al., 2007; Wang and Qian, 2009). Barcoding of the amplicons from each sample site was done using Multiplex Identifier Tags thereby facilitating the assignment of each sequence generated to the correct sample site. PCR amplification of the 16S rRNA gene fragments from each sample was carried out as follows: a 25 µℓ PCR mix consisting of 0.1-1 µℓ of the extracted genomic DNA, 1X PCR buffer (containing MgCl2), 300 µM dNTPs, 0.3 µM of each template specific primer and 0.5 units KAPAHiFi Hotstart DNA Polymerase (KAPA Biosystems) was subjected to initial enzyme activation and DNA denaturation at 95ºC for 5 min followed by cycling parameters of 98ºC for 45 s, 45ºC for 30 s, 72ºC for 45 s (for 5 cycles) and 98ºC for 45 s, 50ºC for 30 s and 72ºC for 45 s (10 cycles). A final extension was done at 72ºC for 5 min. The resultant 468 nt PCR products were gel purified using the Zymo Gel DNA Recovery kit (Zymo Research). Approximately 2 ng of the PCR product was then subjected to a second PCR amplification using FLX fusion primers (consisting of sequencer specific nucleotides, multiplex identifier tag and template-specific nucleotides), as described below. Each sample was amplified with a primer set containing a different multiplex tag. The secondary PCR amplification consisted of 2 ng template, 0.4 µM forward and reverse primers, 300 µM dNTPs, 1X PCR Buffer, 0.5 units KAPAHiFi Hotstart DNA Polymerase (KAPA Biosystems), in a final volume of 25 µℓ. Initial enzyme activation and DNA denaturation was done at 95ºC for 5 min, followed by cycling parameters of 98ºC for 20 s, 52ºC for 45 s, 72ºC for 1 min (for 5 cycles) and 98ºC for 20 s, 65ºC for 45 s and 72ºC for 1 min (15 cycles). A final extension was done at 72ºC for 5 min. The resultant 538 nt PCR products were gel purified as before, the respective amplicons pooled in equal amounts and subjected to emulsion PCR before sequencing using the GS FLX Titanium Sequencer (454 Life Sciences, Roche).
Phylogenetic analysis of rRNA
Sequence reads were quality filtered using the standard software provided by 454 Life Sciences and cured of primer/tag sequences using the CLC Genomics Workbench (CLC Bio). All reads shorter than 100 nt in length were removed from the dataset. The remaining 27 232 reads were analysed using the Ribosomal Database Project (RDP) Classifier, which assigns 16S rRNA sequences to phylogenetic taxonomic groups down to the genus level using a Naïve Bayesian rRNA classifier algorithm (Cole et al., 2009; Wang et al., 2007). The default bootstrap confidence threshold was applied to all sequence reads longer than 250 nt. For reads <250 nt a bootstrap cut-off of 50%, which has been shown to be sufficient to accurately classify sequences at the genus level when V4 regions of the 16S rRNA are targeted (Claesson et al., 2009), was applied. The rarefaction calculations were carried out using the rarefaction analysis tool on the RDP Pyrosequencing Pipeline and curves generated for 0.01, 0.03, 0.05 and 0.1 distance values (Cole et al., 2009).
Results
Physico-chemical and biological variables
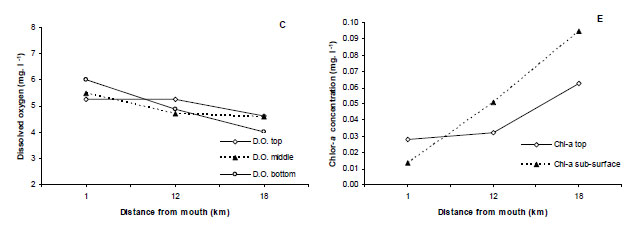
Distinct horizontal gradients in water temperature, salinity and dissolved oxygen concentration were observed within the Kariega Estuary during the study. Water temperatures ranged from 20.3ºC to 17.5ºC, with the highest temperatures recorded in the upper reaches of the estuary (Fig. 2A). Indeed, water temperatures in the upper reaches were significantly higher than those recorded in the lower reaches of the system (F 2,6 = 715, p < 0.001). There were no significant vertical patterns in water temperature evident (p> 0.05). Hypersaline conditions (420/00) prevailed in the upper reaches and marine waters (350/00) in the lower reaches of the estuary (Fig. 2B). Again, the salinity values recorded in the upper reaches of the estuary were significantly higher than those recorded at the mouth of the system (F 2,6 = 361, p < 0.001). There was no significant difference in the salinity values at the different water levels (p > 0.05). Dissolved oxygen (DO) concentrations ranged from 3.76 to 6.02 mg.ℓ-1. There was a significant difference in the levels of DO between the sites (F 2,6 = 9.1, p = < 0.05) with the highest levels being recorded at the mouth of the estuary. There were no significant vertical patterns in DO concentrations evident at each station (p > 0.05) (Fig. 2C).
No significant vertical or horizontal patterns in POM concentrations were observed during the study (F 2,6, 10 = 2.957, p = 0.116; F 2,66 = 2.406, p > 0.05). POM concentrations ranged from 35.80 to 50.40 mg·ℓ-1 in the estuary (Fig. 2D). Total chl-a concentrations ranged from 0.032 to 0.063 mg·ℓ-1 in the surface waters and from 0.060 to 0.104 mg·ℓ-1 at sub-surface levels (Fig. 2E). Again, there were no significant vertical or horizontal patterns in total chl-a concentration evident during the investigation (F 2,6 = 1.317, F 2, 6 = 1.145, p > 0.05 in both cases).
Bacterial diversity
A total of 29 524 16S rRNA sequence reads were generated by the pyrosequencing of duplicate samples from 3 sites (Station 1, 2 and 3) in the estuary. Subsequent to sequence quality control, primer trimming and size exclusion, a sum of 27 232 reads remained with 6 114, 7 913, and 13 205 assigned to Stations 1 (river head), 2 (middle reaches), and 3 (river mouth). These reads were classified, using the RDP 16S rRNA database, down to the level of genus where possible. The relative proportions of bacterial phylotypes in each of the duplicate samples were consistent with respect to the comparative trends in phylogenetic diversity observed between the upper and lower stations. While some variation was observed between the duplicates at Station 2, this did not affect the overall trends observed. Consequently, the 2 data sets generated for each station were pooled for all subsequent analyses.
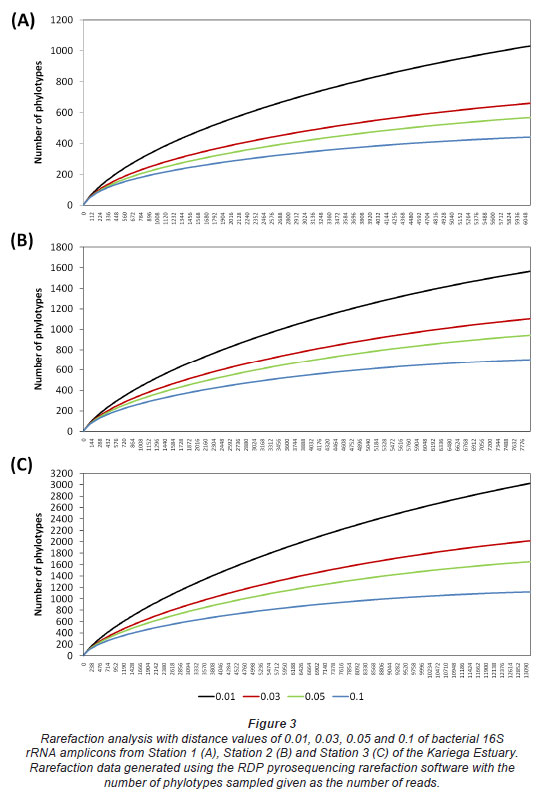
Rarefaction analysis was employed to standardise and compare observed taxon richness between samples and to identify sites that were unequally sampled. Within rarefaction curves, distance values of 0.03, 0.05, and 0.1 are generally accepted as points at which differentiation occurs at the species, genus and family/class level, respectively (Stackebrandt and Goebel, 1994; Hugenholtz et al., 1998; Sait et al., 2002). As there is some debate about these distinctions, particularly the 0.03 cut-off for novel isolates, a more stringent 0.01 distance value was also calculated for the rarefaction curve (Fig. 3). A rarefaction curve which reaches a plateau reflects a habitat that has been sampled to saturation with respect to species diversity in that ecosystem (Hughes and Hellmann, 2005). Our analyses showed that all 3 stations along the length of the Kariega Estuary had been sampled almost to completion and thus the reads analysed for each station were an accurate representation of the bacterial diversity present at the 3 stations during the study (Fig. 3). This result supports the usefulness of pyrosequencing as a tool for deep coverage of bacterial diversity as a function of 16S rRNA gene sequences in aquatic ecosystems. The rarefaction curves also provide comparisons of relative diversity between the upper, middle and lower reaches of the Kariega Estuary. Taking the x-axis values into account (Fig. 3), relative richness of species diversity for the samples processed from the upper reaches of the Kariega Estuary was significantly lower than the bacterial richness found at the estuary mouth.
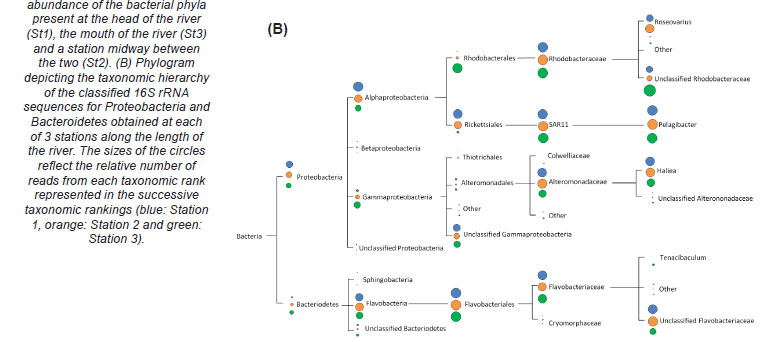
The majority of the bacteria present along the length of the Kariega Estuary belonged to the phyla Proteobacteria and Bacteroidetes (Fig. 4A). It is interesting to note that the relative abundances of reads classified as Bacteroidetes formed a gradient along the river with the concentrations of Bacteroidetes increasing from 15.8% (head of river) to 39% as one progresses down toward the mouth of the estuary (Fig. 4A). The bacterial diversity within the phylum Bacteroidetes present within the Kariega River system consisted almost exclusively of bacteria belonging to the family Flavobacteriaceae (Fig. 4B). A closer investigation of the dominant phylum, namely Proteobacteria, revealed that the majority of the representative reads in this phylum fell within the classes Alphaproteobacteria and Gammaproteobacteria, which respectively form a decreasing and increasing gradient from the river head to the mouth (Fig. 4B). The variance in bacterial phylotype occurrence within the phylum Proteobacteria, between the upper and lower reaches of the river, was further emphasised by the relative dominance of the order Rhodobacterales over Rickettsiales in the upper reaches, with the opposite being true for the lower reaches of the river. With respect to the lower taxon classification of Gammaproteobacteria, the majority of the reads were unclassified (Fig. 4B). An important caveat to note is that the 16S rRNA gene amplicons were generated by PCR and, while the number of cycles was kept to a minimum, the exponential amplification of the more abundant template sequences would surpass that of the low-abundance sequences. This does not affect the identification of dominant phyla but it must be kept in mind that the relative percentages of the dominant versus the less well-represented phyla may be exaggerated.
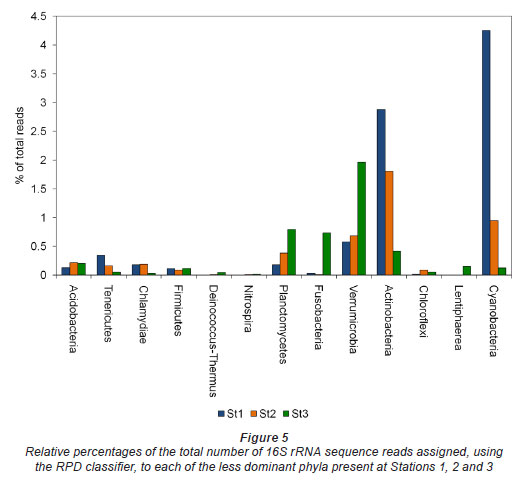
The spatial distribution of the less prominent phyla also demonstrated a strong horizontal pattern (Fig. 5). At the river head, the most abundant bacterial species fell within the phyla Cyanobacteria and Actinobacteria. By contrast, the station at the mouth of the estuary contained a greater representation of microbes from the phyla Verrucomicrobia, Fusobacteria and Planctomycetes with a much lower number of Actinobacteria and Cyanobacteria present. The station occupied in the middle reaches of the estuary comprised bacterial phylotypes from both the upper and lower reaches of the estuary. A small proportion of the reads from each point along the river (2% to 6%, Fig. 4A) did not show significant similarity to any of the 1 379 424 sequences deposited on the Ribosomal Database Project (RDP) database (as of July 2010) and were thus grouped as unclassified bacteria. This is not unexpected as the microbial diversity present in nature is not yet completely characterised and significant numbers of unique bacterial phylotypes have yet to be discovered.
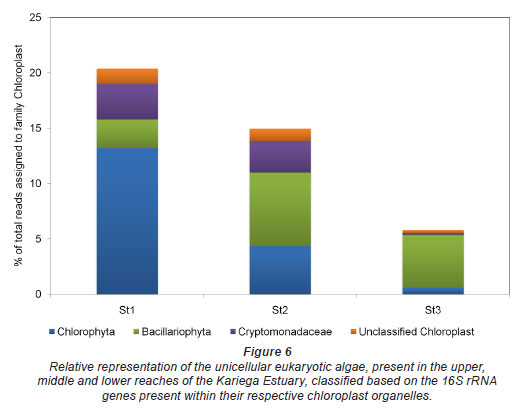
Chloroplasts are the photosynthetic factories within phototrophic eukaryotes, including aquatic unicellular and multicellular algae. Based on similarity in size, morphology and genetic material, chloroplasts are thought to be descendents of a cyanobacterial endosymbiont (Cavalier-Smith, 2002). These organelles contain genes encoding for 16S, 5S, and 23S rRNA, which are phylogenetically most closely related to cyanobacteria (Harris et al., 1994). In the context of this study it is therefore to be expected that the filtering process would net unicellular eukaryotic organisms in addition to prokaryotes. The universal primers for 16S rRNA amplification would consequently amplify the 16S rRNA genes found within the chloroplasts of these organisms as well. Classification of these 16S rRNA gene sequences revealed that most of the photosynthetic unicellular eukaryotes in the Kariega Estuary were found in the upper reaches of the system (Fig. 6). Furthermore, of the chloroplast 16S rRNA genes sequenced, Bacillariophyta (diatoms) dominated at the river mouth whilst Chlorophyta dominated upstream.
Overall, it was evident that the upper and lower reaches of the Kariega Estuary system exhibit clear variations in the distribution of bacterial phylotypes (Figs. 4 and 5), reflecting the contrasting physico-chemical parameters recorded at these stations (Fig. 2). Station 2, in comparison, reveals an intermediate bacterial representation profile to those found at the flanking stations (Figs. 4 and 5). This reflects the physico-chemical properties of the water column mid-way along the river, relative to the head-waters and estuary mouth (Fig. 2).
An important consideration which is readily addressed in this study is the potential occurrence of waterborne pathogens within the Kariega Estuary. Well characterised and emerging bacterial waterborne pathogens include Enterobacteriaceae, Yersinia enterocolitica, Campylobacter jejuni, Vibrio cholera, Mycobacterium spp., Legionella pneumophila, Pseudomonas aeruginosa, Aeromonas spp., Shigella and Salmonella spp. (Leclerc et al., 2002; Sharma et al., 2003). With the exception of Vibrio, none of the above pathogens were detected at the genus level within the 27 232 16S rRNA sequence reads generated during this study. A total of 56 reads (0.002% of the total number of reads) were assigned to the Vibrio genus, using a confidence threshold of 80%. This genus, however, encompasses a diverse group of heterotrophic, ubiquitous marine bacteria, including many facultative symbiotic strains (Thompson et al., 2004). Moreover, bioinformatic analysis of these 56 sequences indicated highest sequence identities to Vibrio species other than Vibrio cholera.
In addition to the above-mentioned pathogens, several cyanobacterial species are able to synthesise a wide range of toxins, which represents a major health risk. These toxins include hepatotoxins (microcystins, nodularins and cylindrospermopsins), neurotoxins (anatoxins and saxitixins) and dermatotoxins, which are produced by several genera within the phylum Cyanobacteria (for details see Briand et al., 2003). None of these potentially toxic cyanobacteria were detected in the Kariega Estuary. In summation, due to the acute sampling depth achieved in this study as indicated by the rarefaction analysis (Fig. 3), the absence of pathogenic bacteria from the Kariega Estuary is clearly apparent.
Discussion
In agreement with the published literature, the permanently-open Kariega Estuary demonstrated a reverse gradient in salinity and temperature (Froneman, 2000; 2001; Heyns and Froneman, 2010). The observed pattern can largely be attributed to, amongst others, a small catchment size (≈ 680 km2) and reduced freshwater inflow into the estuary as a result of several impoundments along the Kariega River (Allanson and Read, 1987; Grange and Allanson, 1995; Grange et al., 2000; Froneman, 2000). Freshwater inflow into the estuary was further reduced by the severe drought within the region during the survey. The limited freshwater inflow into the estuary and generally shallow depth, coupled with strong coastal winds, likely contributed to the virtual absence of any vertical patterns in selected physico-chemical and biological variables during the investigation (Fig. 2).
The composition of a given bacterial community in any specific environment is dependent on the interplay between various factors, including the availability of required nutrients (substrates) and the mortality rates due to biological and/or physical factors (Dolan, 2005). Of the physico-chemical factors present in ecosystems, salinity has been identified as the major environmental determinant of microbial community composition (Lozupone and Knight, 2007). A shift in the gross composition of bacterial communities has been shown along salinity gradients between saline (marine) and freshwater aquatic ecosystems (Crump et al., 1999; Bouvier and del Giorgio, 2002; Kirchman et al., 2005; Lozupone and Knight, 2007). This trend is also reflected in the salinity gradient between saline and hypersaline ecosystems in the Kariega Estuary. Furthermore, the more extreme hypersaline environment present in the upper reaches of the Kariega Estuary are characterised by decreased bacterial community diversity. This reduction of phylotype diversity in response to extreme environments holds true in most instances, except with respect to the bacterial diversities observed in the Tibetan plateau lakes (Wu et al., 2006), where diversity was determined using denaturing gradient gel electrophoresis (DGGE) analysis. This method is far from accurate for 2 reasons:
• Bacterial species encoding 16S rRNA amplicons with similar GC contents will not be distinguishable from one another during DGGE analysis
• Only the dominant phylotypes present in the ecosystem will be detected thus leaving the rare biosphere uncharacterised
Both of these drawbacks are circumvented by the use of pyrosequencing technologies.
The importance of freshwater inflow in promoting phytoplankton production in estuaries, both locally and internationally, is well known (see Adams and Bate, 1999). The estimates of total chl-a concentration recorded during this study (range 0.04 to 0.10 mg·ℓ-1) are in the range reported for the estuary by Froneman (2002) and Heyns and Froneman (2010). The concentrations are, however, substantially lower than those recorded in estuaries within the same geographic region with sustained freshwater inflow (see for example Adams and Bate, 1999). In these systems, total chl-a concentrations can attain levels of up to 200 mg·ℓ-1 with values >10 mg·ℓ-1 not uncommon. The low chl-a concentrations recorded in the permanently open Kariega Estuary can be ascribed to reduced phytoplankton growth rates conferred by low macronutrient availability as a result of the low freshwater inflow (Adams and Bate, 1999). Of the phytoplankton that are present, as indicated by the chloroplast 16S rRNA, Chlorophyta dominated in the upper reaches whilst diatoms (Bacillariophyta) were more prevalent at the mouth of the estuary. The occurrence of cyanobacteria in the Kariega Estuary, particularly in the upper reaches, further contributes to the primary productivity of this ecosystem. It should be noted, however, that chlorophyll-based phototrophs may not be solely responsible for primary productivity in aquatic ecosystems (DeLong and Béjá, 2010). Light-dependent proton pumps in the form of proteorhodopsins have been identified in Pelagibacter ubique from the SAR11 clade (Giovannoni et al., 2005) and have subsequently been found in Pelagibacter spp., Vibrio spp., and Flavobacteria isolates. Furthermore, based on data from ocean genome surveys, an estimated 13% to 80% of marine bacteria express this protein (DeLong and Béjá, 2010). While the metabolic role of proteorhodopsin has not been fully characterised, studies of marine Vibrio cells showed increased starvation-survival in light compared to dark conditions, indicating that proteorhodopsin-containing bacteria may substantially add to the list of phototrophs present in surface aquatic ecosystems (Gómez-Consarnau et al., 2010; DeLong and Béjá, 2010). It is postulated that these light-dependent proteorhodopsin proton pumps play an important role in primary productivity in the oceans by supplying energy for microbial metabolism (Giovannoni et al., 2005; DeLong and Béjá, 2010). Both Pelagibacter spp. (phylum Alphaproteobacterium) and Flavobacteria (phylum Bacteroidetes) occurred throughout the Kariega Estuary in significant numbers.
The two most well represented phyla in the Kariega Estuary were the Proteobacteria and Bacteroidetes, which exhibit reverse trends in their spatial distribution (Fig. 4A). The Proteobacteria were comprised almost exclusively of Alphaproteobacteria and Gammaproteobacteria. Both these taxa have been previously detected in hypersaline environments (Wu et al., 2006). Alphaproteobacteria occur in higher densities as salinity increases (Benlloch et al., 2002; Bouvier and Del Giorgio, 2002; Kirchman et al. 2005; Wu et al. 2006), which corresponds well with the observed increase in Alphaproteobacteria towards the hypersaline upper reaches of the Kariega Estuary during the present study. The relative abundances of Gammaproteobacteria in the hypersaline regions of the Kariega Estuary were lower than those recorded at the mouth of the system. This contrasts with the high percentages of Gammaproteobacteria reported under hypersaline conditions (Benlloch et al., 2002; Humayoun et al., 2003; Wu et al., 2006, Demergasso et al., 2008) but not in the polysaline conditions present in estuaries (Bouvier and Del Giorgio, 2002; Kirchman et al., 2005). However, the majority of the Gammaproteobacteria present in the Kariega Estuary did not show significant similarity to any known isolate and may represent a specialised subgroup of bacteria adapted to this particular environment.
The second-most abundant bacterial phylum in the Kariega Estuary was the Bacteroidetes, which is also referred to as the CFB (Cytophaga-Flavobacteria-Bacteroides) division of bacteria. Bacteroidetes occur in marine environments, often as the dominant phylotype (Cottrell and Kirchman, 2000b; Eilers et al., 2001) and are found as free-living organisms or associated with organic aggregates, phytoplankton and marine animals (DeLong et al., 1993; Eilers et al., 2001; Webster et al., 2001; Abell and Bowman, 2005; Grossart et al., 2005). This group of bacteria are chemo-organotrophic and are especially proficient in the uptake and degradation of complex dissolved and particulate organic matter suggesting that they play an important role in the detritus food chain and carbon cycling in aquatic ecosystems (Reichenbach, 1992; Cottrell and Kirchman, 2000a; Kirchman, 2002). Based on the ability of different bacterial groups to utilize dissolved organic matter (DOM), it is suggested that Bacteroidetes in combination with Alpha- and Gammaproteobacteria are responsible for the uptake and mineralization of DOM (Cottrell and Kirchman, 2000a). The numerical dominance of these groups within the bacterial system of the Kariega Estuary highlights the importance of the detrital food web within the system. This is further supported by the presence of the less well-represented bacterial phyla Actinobacteria and Planctomycetes which are indicated as important players in the mineralisation of DOM and POM in aquatic ecosystems (Bull et al., 2005; Gade et al., 2004). These results are in agreement with a previous study which suggested that the microbial loop predominates within the freshwater-deprived Kariega Estuary ecosystem (Froneman and McQuaid, 1997).
Of critical importance when considering the composition of the bacterial community within the Kariega Estuary is the potential impact of human pollution on the system. One of the most easily identifiable markers of human pollution is the presence of pathogenic microorganisms, many of which originate from untreated or poorly treated sewage and rainwater runoff from unhygienic locations. Sampling of the bacterial population along the length of the Kariega Estuary was done to such a degree that the presence of pathogenic isolates in any significant numbers at all would have been detected. The Kariega Estuary is considered to be in a good ecological state based on geomorphology, ichthyofaunal community structure and aesthetics (Harrison et al., 2000) and this is substantiated by the absence of pathogenic bacteria.
Conclusions
This study was a pilot project to investigate the suitability of using pyrosequencing as a tool for characterising the bacterial diversity in an oligotrophic permanently-open freshwater-deprived Eastern Cape estuary. Although it is not feasible to elucidate ecosystem function based solely on phylogeny, the presence of distinct prokaryotic lineages within the Kariega Estuary clearly highlights the importance of the detrital food web with a less dominant, supporting, bacterial primary productivity. A distinct horizontal variation in phylotype representation was observed in the bacterial community, reflecting the reverse salinity gradient present in the estuary. While PCR amplification negates the capability to define the absolute numbers for each taxonomic unit present in the samples, pyrosequencing of 16S rRNA genes does provide a reliable reflection of the relative abundances of the respective phylotypes. Rarefaction analysis of the sequence data generated in this study indicates that bacterial sampling at each station was close to saturation levels, thereby verifying that the overwhelming majority of bacterial phylotypes present at each point along the Kariega Estuary were detected. The absence of 16S rRNA sequences representing human pathogenic microbes in the water samples highlights the good ecological state of this system. Furthermore, by altering the primer set used in the amplification cycle to primers specific for the 18S rRNA gene, sampling of unicellular fungal and eukaryotic organisms may be easily achieved. We conclude that pyrosequencing has provided a versatile and efficient tool for assessing the bacterial diversity in the Kariega Estuary and propose that this technology may provide valuable information in assessing the health of both freshwater and marine ecosystems.
Acknowledgments
Gwynneth Matcher was supported by an NRF Freestanding Innovation Postdoctoral Fellowship. We would like to thank Rhodes University for providing funds and facilities for this study.
References
ABELL GCJ and BOWMAN JP (2005) Colonization and community dynamics of class Flavobacteria on diatom detritus in experimental mesocosms based on Southern Ocean seawater. FEMS Microbiol. Ecol. 53 379-391. [ Links ]
ADAMS JB and BATE GC (1999) Primary producers. Estuarine microalgae. In: Allanson BR and Baird D (eds.) Estuaries of South Africa (1st edn.). Cambridge University Press, Cambridge. 91-117. [ Links ]
ALLANSON BR and READ GHL (1987) The response of estuaries along the south eastern coast of South Africa to marked variation in freshwater inflow. Special Report No. 2/87. Institute for Freshwater Studies, Rhodes University, Grahamstown. [ Links ]
BENLLOCH S, LÓPEZ-LÓPEZ A, CASAMAYOR EO, ØVREAS L, GODDARD V, DAAE FL, SMERDON G, MASSANA R, JOINT I, THINGSTAD F, PEDRÓS-ALIÓ C and RODRÍGUEZ-VALERA F (2002) Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ. Microbiol. 4(6)349-360. [ Links ]
BOUVIER TC and DEL GIORGIO PA (2002) Compositional changes in free-living bacterial communities along a salinity gradient in two temperate estuaries. Limnol. Oceanogr. 47(2)453-470. [ Links ]
BRIAND J, JACQUET S, BERNARD C and HUMBERT J (2003) Health hazards for terrestrial vertebrates from toxic cyanobacteria in surface water ecosystems. Vet. Res. 34 361-377. [ Links ]
BULL AT, STACH JEM, WARD AC and GOODFELLOW M (2005) Marine actinobacteria: perspectives, challenges, future directions. Antonie van Leeuwenhoek 87 65-79. [ Links ]
CAVALIER-SMITH T (2002) Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 12 R62-R64. [ Links ]
CLAESSON MJ, O'SULLIVAN O, WANG Q, NIKKILA J, MARCHESI JR, SMIDT H, DE VOS WM, ROSS RP and O'TOOLE PW (2009) Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 4(8)e6669. doi: 10.1371/journal.pone.0006669. [ Links ]
COLE JR, WANG Q, CARDENAS E, FISH J, CHAI B, FARRIS RJ, KULAM-SYED-MOHIDEEN AS, McGARRELL DM, MARSH T, GARRITY GM and TIEDJE JM (2009) The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. NAR. 37 (Database Issue) D141-D145. URL: http://rdp.cme.msu.edu/classifier/classifier.jsp; http://pyro.cme.msu.edu/. [ Links ]
COTTRELL MT and KIRCHMAN DL (2000a) Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl. Environ. Microbiol. 66(4)1692-1697. [ Links ]
COTTRELL MT and KIRCHMAN DL (2000b) Community composition of marine bacterioplankton determined by 16S rRNA gene clone libraries and fluorescence in situ hybridization. Appl. Environ. Microbiol. 66 5116-5122. [ Links ]
CRUMP BC, ARMBRUST EV and BAROSS JA (1999) Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia River, its estuary, and the adjacent coastal ocean. Appl. Environ. Microbiol. 65 3192-3204. [ Links ]
DELONG EF, FRANKS DG and ALLDREDGE AL (1993) Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 38 924- 934. [ Links ]
DELONG EF and BÉJÀ O (2010) The light-driven proton pump proteorhodopsin enhances bacterial survival during tough times. PloS Biol. 8(4)e1000359. [ Links ]
DEMERGASSO C, ESCUDERO L, CASAMAYOR EO, CHONG G, BALAGUÉ V and PEDRÓS-ALIÓ C (2008) Novelty and spacio-temporal heterogeneity in the bacterial diversity of hypersaline lake Tebenquiche (Salar de Atacama). Extremophiles 12 491-504. [ Links ]
DOLAN JR (2005) An introduction to the biogeography of aquatic microbes. Aquat. Microb. Ecol. 41 39-48. [ Links ]
EILERS H, PERNTHALER J, PEPLIES J, GLÖCKNER FO, GERDTS G and AMANN R (2001) Isolation of novel pelagic bacteria from the German Bight and their seasonal contribution to surface picoplankton. Appl. Environ. Microbiol. 67 5134-5142. [ Links ]
FORD TE (2000) Response of microbial communities to anthropogenic stress. J. Aquat. Ecosyst. Stress Recovery 7 75-89. [ Links ]
FRONEMAN PW (2000) Preliminary study of the food web structure of two contrasting estuaries along the Eastern Cape coast, South Africa. Afr. J. Aquat. Sci. 25 13-22. [ Links ]
FRONEMAN PW (2001) Seasonal changes in zooplankton biomass and grazing in a temperate estuary, South Africa. Estuarine Coastal Shelf Sci. 52 543-553. [ Links ]
FRONEMAN PW (2002) Food web structure in three contrasting estuaries determined using stable isotope (δ13C) analysis. Afr. J. Aquat. Sci. 27 107-115. [ Links ]
FRONEMAN PW and McQUAID CD (1997) Preliminary investigation of the ecological role of microzooplankton in the Kariega Estuary, South Africa. Estuarine Coastal Shelf Sci. 45 689-695. [ Links ]
GADE D, SCHLESNER H, GLÖCKNER FO, AMANN R, PFEIFFER S and THOMM M (2004) Identification of Planctomycetes with order-, genus-, and strain-specific 16S rRNA-targeted probes. Microb. Ecol. 47 243-251. [ Links ]
GIOVANNONI SJ, BIBBS L, Cho J, STAPELS MD, DESIDERIO R, VERGIN KL, RAPPÉ MS, LANEY S, WILHELM LJ, TRIPP HJ, MATHUR EJ and BAROFSKY DF (2005) Proteorhodopsin in the ubiquitous marine bacterium SAR11. Nature 438 82-85. [ Links ]
GÓMEZ-CONSARNAU L, AKRAM N, LINDELL K, PEDERSEN A, NEUTZE R, MILTON DL, GONZÁLEZJM and PINHASSI J (2010) Proteorhodopsin phototrophy promotes survival of marine bacteria during starvation. PLoS Biol. 8(4)e1000358. [ Links ]
GRANGE N and ALLANSON BR (1995) The influence of freshwater inflow on the nature, amount and distribution of seston in estuaries of the Eastern Cape, South Africa. Estuarine, Coastal Shelf Sci. 40 403-420. [ Links ]
GRANGE N, WHITFIELD AK, DE VILLIERS CJ and ALLANSON BR (2000) The response of South African east coast estuaries to altered river flow regimes. Aquat. Conservation: Mar. Freshwater Ecosyst. 10 155-177. [ Links ]
GROSSART HP, LEVOLD F, ALLGAIER M, SIMON M and BRINKHOFF T (2005) Marine diatom species harbour distinct bacterial communities. Environ. Microbiol. 7 860-873. [ Links ]
HARRIS EH, BOYNTON JE and GILLHAM NW (1994) Chloroplast ribosomes and protein synthesis. Microb. Rev. 58(4)700-754. [ Links ]
HARRISON TD, COOPER JAG and RAMM AEL (2000) State of South African estuaries; geomorphology, icthyofauna, water quality and aesthetics. State of the Environment Series, Report No. 2. Department of Environmental Affairs and Tourism, Pretoria. [ Links ]
HEYNS E and FRONEMAN W (2010) Spatial and temporal patterns in the hyperbenthic community structure in a warm temperate southern African permanently open estuary. Estuarine Coastal Shelf Sci. 88 105-115. [ Links ]
HODGSON AN (1987) Distribution and abundance of macrobenthic fauna of the Kariega Estuary. S. Afr. J. Zool. 22 153-162. [ Links ]
HOLM-HANSEN O and RIEMANN B (1978) Chlorophyll a determination: improvements in methodology. Oikos 30 438-447. [ Links ]
HORNER-DEVINE MC, LEIBOLD MA, SMITH VH and BOHANNAN BJM (2003) Bacterial diversity patterns along a gradient of primary productivity. Ecol. Lett. 6 613-622. [ Links ]
HUGENHOLTZ P, GOEBEL BM and PACE NR (1998) Impact of culture independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180 4765-4774. [ Links ]
HUGHES JB and HELLMANN JJ (2005) The application of rarefaction techniques to molecular inventories of microbial diversity. In: Leadbetter JE (ed.) Methods in Enzymology 397 292-308. [ Links ]
HUMAYOUN SB, BANO N, and HOLLIBAUGH JT (2003) Depth distribution of microbial diversity in Mono Lake, a meromictic soda lake in California. Appl. Environ. Microbiol. 69(2)1030-1042. [ Links ]
HUSE SM, DETHLEFSEN L, HUBER JA, WELCH DM, RELMAN DA and SOGIN ML (2008) Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4(11)e1000255. [ Links ]
JENNINGS ME (2006) Nutrient dynamics in and offshore of two permanently open South African estuaries with contrasting freshwater inflow. MSc thesis, Rhodes University. [ Links ]
KIRCHMAN DL (2002) The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39 91-100. [ Links ]
KIRCHMAN DL, DITTEL AI, MALMSTROM RR and COTTRELL MT (2005) Biogeography of major bacterial groups in the Delaware Estuary. Limnol. Oceanogr. 50(5)1697-1706. [ Links ]
KIRCHMAN DL (2008) New light on an important microbe in the ocean. Proc. Natl. Acad. Sci. 105(25)8487-8488. [ Links ]
LECLERC H, SCHWARTZBROD L and DEI-CAS E (2002) Microbial agents associated with waterborne diseases. Crit. Rev. Microbiol. 28(4)371-409. [ Links ]
LEMKE MJ, BROWN BJ and LEFF LG (1997) The response of three bacterial populations to pollution in a stream. Microb. Ecol. 34 224-231. [ Links ]
LOZUPONE CA and KNIGHT R (2007) Global patterns in bacterial diversity. PNAS 104(27)11436-11440. [ Links ]
LIU Z, LOZUPONE C, HAMADY M, BUSHMAN FD and KNIGHT R (2007) Short pyrosequencing reads suffice for accurate microbial community analysis. NAR 35(18)e120. [ Links ]
REICHENBACH H (1992) The Order Cytophagales. In: Balows A, Trüper HG, Dworkin M, Harder W and Schleifer KH (eds.) The Prokaryotes (2nd edn.) Vol. 4. Springer, Berlin. 3631-3687. [ Links ]
SAIT M, HUGENHOLTZ P and JANSSEN PH (2002) Cultivation of globally distributed soil bacteria from phylogenetic lineages previously only detected in cultivation-independent surveys. Environ. Microbiol. 4 654-666. [ Links ]
SCHLOSS PD and HANDELSMAN J (2005) Metagenomics for studying unculturable microorganisms: Cutting the Gordian knot. Genome Biol. 6(8)229 [ Links ]
SHARMA S, SACHDEVA P and VIRDI JS (2003) Emerging water-borne pathogens. Appl. Microbiol. Biotechnol. 61 424-428. [ Links ]
SMITH VH (2007) Microbial diversity productivity relationships in aquatic ecosystems. FEMS Microbiol. Ecol. 62 181-186. [ Links ]
SOGIN ML, MORRISON HG, HUBER JA, WELCH DM, HUSE SM, NEAL PR, ARRIETA JM and HERNDL GJ (2006) Microbial diversity in the deep sea and the unexplored 'rare biosphere'. Proc. Natl. Acad. Sci. U.S.A. 103 12115-12120. [ Links ]
STACKEBRANDT E and GOEBEL BM (1994) A place for DNA-DNA reassociation and 16S rRNA sequence-analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44 846-849. [ Links ]
TEIRA E, GASOL JM,ARANGUREN-GASSIS M, FERNANDEZ A, GONZALEZ J, LEKUNBERRI I and ALVAREZ-SALGADO XA (2008) Linkages between bacterioplankton community composition, heterotrophic carbon cycling and environmental condition in a highly dynamic coastal system. Environ. Microbiol. 10(4)906-917. [ Links ]
THOMPSON JR, RANDA MA, MARCELINO LA, TOMITA-MITCHELL A, LIM E and POLZ MF (2004) Diversity and dynamics of North Atlantic coastal Vibrio community. Appl. Environ. Microbiol. 70 (7) 4103-4110. [ Links ]
TRINGE SG and HUGENHOLTZ P (2008) A renaissance for the pioneering 16S rRNA gene. Curr. Opin. Microbiol. 11 442-446. [ Links ]
WANG Y and QIAN P (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4(10)e7401. [ Links ]
WANG Q, GARRITY GM, TIEDJE JM, and COLE JR (2007) Naïve Bayesian Classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73(16)5261-7. [ Links ]
WEBSTER NS, WILSON KJ, BLACKALL LL and HILL RT (2001) Phylogenetic diversity of bacteria associated with the marine sponge Rhopaloeides odorabile. Appl. Environ. Microbiol. 67 434-444. [ Links ]
WU QL, ZWART G, SCHAUER M, KAMST-VAN AGTERVELD MP and HAHN MW (2006) Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl. Environ. Microbiol. 72(8)5478-5485. [ Links ]
Received 2 November 2010; accepted in revised form 31 May 2011.
* To whom all correspondence should be addressed. +2746 603 7039; fax: +2746 622 3984; e-mail: g.matcher@ru.ac.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}