










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.37 n.1 Pretoria Jan. 2011
The electro-oxidation of lignin in Sappi Saiccor dissolving pulp mill effluent
B MoodleyI,*; DA MulhollandI,II; HC BrookesI,II
ISchool of Chemistry, University of KwaZulu-Natal, Durban, 4041, South Africa
IIDivision of Chemical Sciences, Faculty of Health and Medical Sciences, University of Surrey, Guildford, GU2 7XH, United Kingdom
ABSTRACT
Electro-oxidation reactions using a nickel anode were carried out on the calcium-spent liquor effluent obtained from Sappi Saiccor (formerly South African Industrial Cellulose Corporation) dissolving pulp mill as well as on lignin- and lignan-type compounds previously identified in the effluent. Voltammograms were obtained for each solution in order to identify the oxidation potentials of the compounds to be electro-oxidised. Value-added products such as vanillin and syringaldehyde were identified in the electro-oxidised reaction mixtures using gas chromatography-mass spectrometry (GC-MS) and high performance liquid chromatography (HPLC). Profiles of the changes in concentration of these compounds were determined as a function of time with maximum concentrations reached within the first half hour. These findings are significant in that few electro-oxidation reactions have been carried out on the effluent of a pulp mill which uses the acid bisulphite pulping process and no results have previously been reported on the electro-oxidation of syringaldehyde. This study contributes to a further understanding of the electro-oxidation of lignin and is of value to the paper and pulp industry at large. Reduction of the organic content of the effluent by electro-oxidation was shown to be possible.
Keywords: electro-oxidation, lignin, effluent, vanillin, syringaldehyde
Introduction
Sappi Saiccor, situated near Durban, South Africa, is one of the few pulp mills that still partially use calcium bisulphite as the base for the acid bisulphite pulping process. This process produces a high-grade cellulose pulp but significant volumes of effluent waste are also generated, which are discharged to sea via a 7 km pipeline. Environmental regulations require that effluent reductions are made and therefore Saiccor has embarked on an investigation of the effluent itself.
Research on the effluent has shown it to contain the lignan isomer syringaresinol, as the major constituent, some lignin-type precursors, fatty acids, sugars and lignosulphonates (Moodley et al., 2003). The aim of this project was to oxidise the effluent to produce value-added products that could be extracted for commercial purposes, thereby reducing the amount of effluent pumped out to sea as waste, as well as investigating the use of electro-oxidation as a means of reducing the organic content of the effluent. This is the first attempt at performing any type of oxidation on Sappi Saiccor's effluent and the results could be of value to the industrial paper and pulp community. Previous work on the oxidation of lignin was based primarily on Kraft pulping effluent, which is alkaline, compared to our acid bisulphite pulping effluent. Furthermore, Saiccor uses hardwoods with a higher syringyl content compared to mills that use softwoods which have higher guaicol content. Thus, the source of the wood and the pulping process used combine to produce an effluent unique to Saiccor. Previous electro-oxidation reactions have been done on pure compounds but not on a mixture of compounds obtained from effluent, as carried out in this work. The results presented in this paper for the electro-oxidation of syringaldehyde are also the first reported results in literature.
Many chemical oxidants have been employed for the oxidation of lignin. Although nitrobenzene is a suitable oxidising agent, high temperatures and pressures are necessary and the formation of nitro derivatives make these oxidations commercially and environmentally undesirable. The use of oxygen as an oxidant also requires high temperatures and pressures, which again makes it an undesirable process. Reactions with hydrogen peroxide tend to form acids and often lead to degradation of the aromatic ring. Thus electrolysis of lignin could be an attractive alternative in the oxidation of lignin and lignosulphonates, as it offers several advantages over the traditional chemical oxidation processes. In particular, electrolysis of lignin and lignosulphonates in basic media can use relatively lower temperatures and pressures compared to reactions with oxygen and nitrobenzene, and there is better control of the reaction, leading to greater selectivity (Varey, 1987; Utley and Smith, 1986; 1987; 1988). Degradation of lignin often produces products such as aldehydes, ketones and acids.
Some of the earliest studies on the electro-oxidation of organic molecules involved phenolics, and it was found that electro-oxidation was comparable to chemical oxidation reactions, forming the aromatic aldehyde and acid (James, 1899; Law, 1906). Electro-oxidation of phenolics and lignin model compounds was investigated with the aim of using the technique as an identification tool, and for the oxidation and synthesis of phenolics and lignin derivatives such as vanillin and syringic acid (Hedenberg and Freiser, 1953; Gaylor et al., 1953; Lund, 1957; Vermillion, 1963; Kovalenko et al., 1973; 1976; 2000; Demin et al., 1980). Various graphite and platinum electrodes were investigated and it was realised fairly early that the nature of the electrode surface played an important role (Hedenberg and Freiser, 1953; Gaylor et al., 1953; 1957; Elving and Krivis, 1958; Morris and Schempf, 1959). Many of these studies observed the formation of a thin surface film, which was thought to be the result of polymerisation; recent studies show a possible application of this in the protection of steel (Vagin et al., 2006). Electro-oxidation of lignin type compounds in base using platinum and nickel electrodes showed the formation of dimers, using starting materials obtained from an acidic media (Pearl, 1952), which has also been noted in the present study.
Smith and Utley showed that a combination of nitroaromatics in an electrolysis reaction of lignin in base required lower temperatures than either reaction on its own (Smith and Utley, 1989). The disadvantage was that nitro derivatives were still present in the end product. A patent by Utley and Smith claimed to produce yields of useful monomers, greater than 10%, by passing an electric current through an alkaline solution of lignin at temperatures above 100ºC with continuous mixing (Utley and Smith, 1988). This method claimed to produce vanillin or partial cleavage products such as dimers, trimers or oligomers of monocyclic species. Studies of the mechanism of electro-oxidative cleavage of dimers have shown that aldehydes such as vanillin and syringaldehyde form as well as carboxylic acids (Pardini et al., 1991). These products all formed at a nickel anode resulting from an oxygenated nickel(III) anode surface (Pardini et al., 1991). Recent research on the combination of photochemical and electrochemical oxidation methods on Kraft lignin showed the production of vanillin and vanillic acid (Tian et al., 2010).
Studies were carried out on the influence of different electrode materials such as Pt, Au, Ni, Cu, DSA-O2 (dimensionally stable anode), PbO2 and boron-doped diamond electrodes, on phenolics and lignin electro-oxidation reactions (Kovalenko et al., 1977; Parpot et al., 2000; Canizares et al., 2002; Iniesta et al., 2001). Early studies on graphite, copper, platinum and nickel anodes showed that platinum and nickel produced higher amounts of aldehyde and a comparison of the nickel and DSA-O2 anodes, at the same high current densities of 750 A/m2, showed that nickel produced a higher concentration of vanillin per unit of charge used (Kovalenko et al., 1977; Parpot et al., 2000).
Current research on the electrochemical oxidation of Kraft lignin has involved the use of a number of different natural and synthetic compounds that may act as redox mediators or enhancers during these electrochemical oxidation reactions (Arzola et al., 2009). The use of modified N-hydroxyphthalimide has also been found to be beneficial in promoting electrochemical oxidation of non-phenolic lignin model compounds in the presence of a laccase mediator (Annunziatini et al., 2005).
Experimental
Pre-treatment procedure
The nickel anode, which forms a nickel(III) peroxide surface, was cleaned by holding the electrode at negative, and thereafter positive, potentials for 30 s each, was rinsed with Millipore water and then placed in an ultrasonic bath for 5 min, followed by cleaning in nitric/sulphuric acid mixtures. A nickel working electrode, platinum counter electrode and a Ag/AgCl reference electrode were connected to a BAS potentiostat CV-27. Figures 1 and 2 show the schematic diagram and the actual electrooxidation setup.
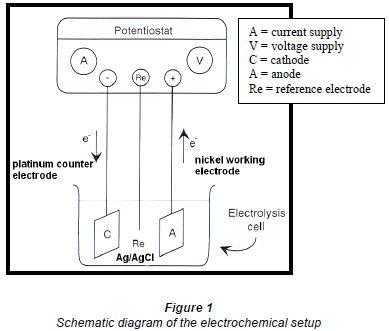

Solution preparation and voltammetry
Standard solutions containing pure syringaldehyde (0.01 M) in KOH (0.1 M) and sodium perchlorate (0.5 M) were prepared in Millipore water. Background voltammograms were obtained for 0.5 g/ℓ solutions of pure compounds in KOH and sodium perchlorate (pH 12.45) with continuous stirring. The solutions were electro-oxidised at the suitable oxidation potentials for a period of time with continuous stirring. The oxidised solution was extracted with chloroform in a sulphuric acid medium. All chemicals used were analytical grade.
Analysis
The end product was analysed using GC-MS and HPLC. GC-MS analysis involved 1 µℓ manual injections onto a HP5-MS column with 1:75 split. The sample was held for 2 min at 50ºC, the temperature was ramped at 20ºC per minute until it reached 300ºC and thereafter the sample was held for 20 min. Low-resolution mass spectrometry was performed on an Agilent 5973 mass spectrometer connected to a 6890GC. The results of the mass spectrum were matched with mass spectra of NIST reference standards.
HPLC analysis was performed on an Agilent system with an UV detector. Filtered samples (25 µℓ in acetonitrile) were introduced onto a ZORBAX C-18 extend column connected to a C-8 column in the LC. The solvent system was increased from 0-100% acetonitrile with 0.2% formic acid in 60 min with a flowrate of 1.000 mℓ/min and the oven temperature was maintained at 40ºC. Some analyses were conducted using a gradient system of 0-100% acetonitrile with 0.2% formic acid in 170 min, with the same flowrate and oven temperature as above but with a stop-time of 60 min and a 10-min flush between samples. Blanks were obtained according to standard procedures. Quantitative analyses were performed using calibration standards with the inclusion of a 0.002 M internal standard, 3, 4 dimethoxybenzaldehyde in a solution of KOH (0.1 M) and NaClO4.H2O (0.5 M), in each standard. For the electro-oxidation reactions, samples (1.5 mℓ) were taken every hour during the electro-oxidation of syringaldehyde and every half hour for the remaining reactions, filtered and placed in vials containing 30 µℓ of filtered 0.1 M internal standard.
The samples were analysed up to 5 times using the LC instrument and concentrations were calculated from the calibration curves. These values were used to calculate the mass of compound produced per gram of starting material. The initial mass of the compound already present per gram of starting material was determined at time = 0 and was subtracted from the mass of compound produced per gram of starting material calculated at each sample time. The resulting mass of compound produced per gram of starting material was converted to a percentage and the average determined. This result is referred to as percentage concentration and will be discussed later.
Results and discussion
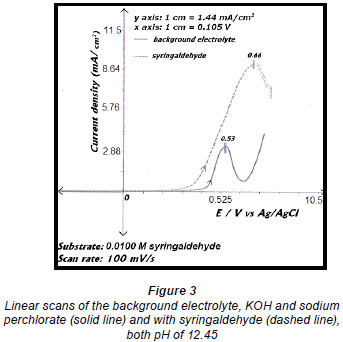
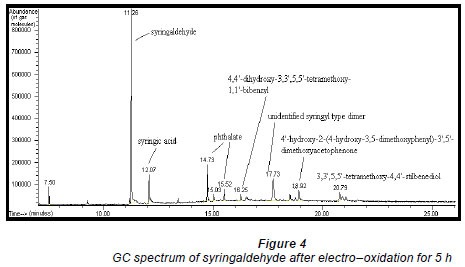
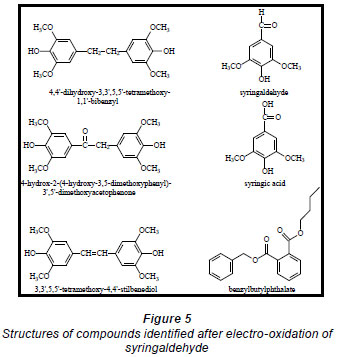
Electro-oxidations were performed on standard solutions of syringaldehyde and the lignan syringaresinol, which are known to be present in the effluent, as well as on the organic-extracted portion of the effluent, a concentrated form of the effluent, and lignosulphonates obtained from Lignotech, all originating from an acid bisulphite pulping process. Cyclic voltammograms (CV) or linear scans were obtained for each solution including the background electrolyte on its own. The linear scans (Fig. 3) for 0.5 g/ℓ of syringaldehyde solution (dashed line) showed an oxidation peak potential at 0.66 V vs. Ag/AgCl compared to the oxidation peak for the background electrolyte on its own (solid line). After electro-oxidation for 5 h at 0.66 V, analysis of the end product showed the presence of syringic acid together with other dimers and compounds (Fig. 4). The presence of syringic acid in the final mixture confirmed that electro-oxidation had occurred. The identification of each peak in the GC spectrum was based on its mass spectrum and the structures of the dimers were confirmed by comparison of the mass spectra with published data (Majumder and Pal, 1993; Bangani, 1998; Pearl, 1957).
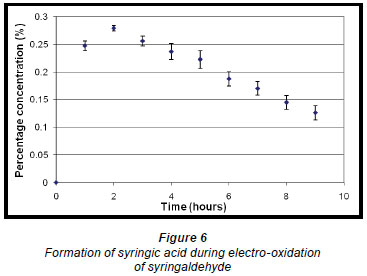
The phthalates identified are possibly from the plastic fittings used during the reaction. A quantitative analysis of the reaction product showed that the maximum conversion of syringaldehyde to syringic acid occurred within the first 2 h of electro-oxidation yielding 0.28% (of starting material) (± 0.48 x 10-2%) of syringic acid (Fig. 6). Thereafter, the percentage concentration of syringic acid decreased suggesting a method of effluent cleaning. In this work, the percentage concentration is the mass percentage concentration of the compound formed from the starting material. It is calculated from the following formula:
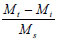
where:
Mt is the mass of the compound at time t,
Mi is the mass of the compound at time 0 and
Ms is the mass of the starting material.
Since the definition of percentage concentration used in this work is based on the additional mass of compound produced per gram of starting material, if the additional mass produced decreases over time due to electro-degradation, the percentage concentration will decrease. Furthermore, the initial starting material (in the case of the concentrated effluent, organic portion of the effluent and the lignosulphonate mixture) already contains a certain mass of the compound of interest which may be destroyed or more may be produced during the reaction. This initial starting material must be subtracted from the mass of compound produced at each sample time to give a true reflection of the actual amount of compound produced from the reaction itself. If the overall mass of compound per gram of starting material present at each sample point decreases and the initial mass of compound present is subtracted, negative concentrations may be obtained because the change in mass of compound produced per gram of starting material is plotted against time, and not the total mass of compound produced per gram of starting material. These reduced values will also be referred to as percentage concentrations in order to maintain consistency and allow for comparison of different compounds.
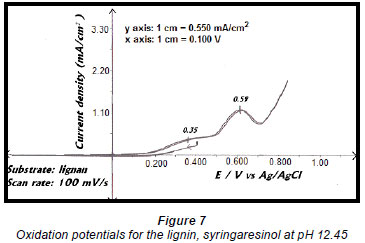
Studies on the effluent identified the lignan syringaresinol as the major component and therefore electro-oxidation reactions were carried out on this compound (Moodley et al., 2003). A linear sweep of a 0.5 g/ℓ solution of the lignan showed oxidation peaks at 0.35 V vs. Ag/AgCl and 0.59 V vs. Ag/AgCl (Fig. 7). This suggested that the oxidation of syringaresinol proceeds via a 2-step process that forms an intermediate, which is then oxidised to the final product (Pearl, 1957; Yoshida, 1984).
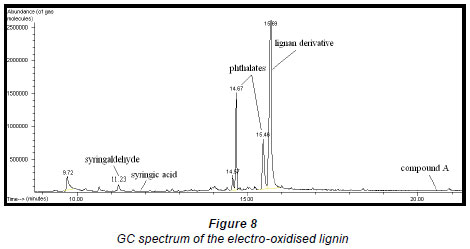
Qualitative analysis of the electrooxidised extract of the lignan by GC-MS (Fig. 8) showed the presence of a lignan derivative, some phthalate contaminants, a small amount of syringaldehyde and an even smaller amount of syringic acid whose peak is not clearly seen in Fig. 8. The structures of Compound A and the lignan derivative were not conclusively identified as insufficient quantities of these compounds were obtained.
The quantitative analysis of syringaldehyde and syringic acid formed during the reaction showed that the concentration of syringaldehyde increased slightly and thereafter stabilised after about 2.5 h.
(Fig. 9). Syringic acid concentration on the other hand decreased. In this case a small amount of syringic acid forms during the pre-treatment stage and is subtracted from syringic acid formed during the reaction. (Pre treatment refers to the dissolution of the oxidisable species in an alkaline solution.) Since the total amount of syringic acid also decreased as the reaction proceeded and together with the very small amount formed initially during the reaction, the eventual change in mass of syringic acid produced per gram of starting material is a 'negative or reduced percentage concentration' that reduces further as the reaction proceeds.
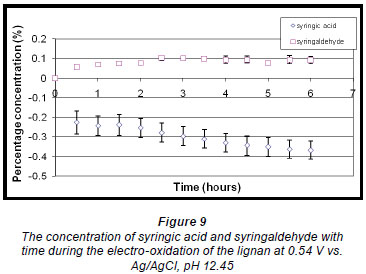
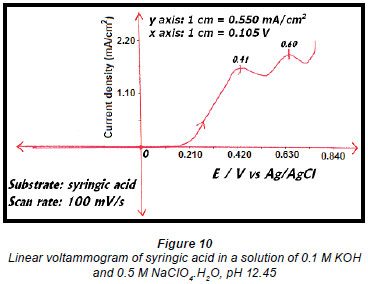
A linear voltammogram of syringic acid showed oxidation peaks at 0.41 V vs. Ag/AgCl and 0.60 V vs. Ag/AgCl (Fig. 10). Since the electro-oxidation of syringaresinol solution occurred at 0.54 V vs. Ag/AgCl, any species with an oxidation potential below that value would also be oxidised. Therefore syringic acid underwent some form of oxidation because its first oxidation potential is less than that of 0.54 V vs. Ag/AgCl, resulting in a decrease in the concentration of syringic acid (Fig. 9).
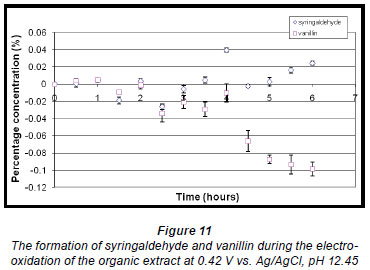
A voltammogram of the organic extract also showed 2 oxidation potentials. Analysis of the electro-oxidation reaction mixture produced at the lower potential showed that the concentration of vanillin decreased over time whereas the concentration of syringaldehyde increased after approximately 4 h (Fig. 11).
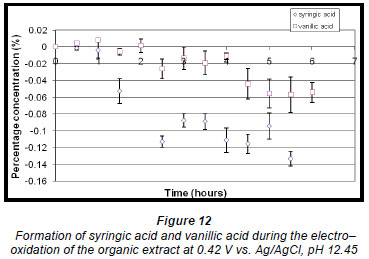
The percentage concentration of vanillic acid increased very slightly to approx. 0.0078% (± 0.2048 x 10-2%) within the first hour but then decreased about half way through the reaction. The concentration of syringic acid decreased throughout the reaction (Fig. 12) because the oxidation potential for this reaction was approximately at the same potential as that of syringic acid (Fig. 10).
Graphs of the percentage concentration vs. time in hours for the above compounds showed that the concentrations of these compounds decreased to values below the starting value during long periods of electro-oxidation. This suggested that the electro-oxidation reactions carried out over long periods of time eventually destroy the compounds that the reaction initially formed. Therefore, a continuous extraction process is required before complete destruction of the products. This was also recommended by Parpot et al., who observed similar results during their studies of vanillin production from the electro-oxidation of lignin obtained via the alkaline Kraft pulping process (Parpot et al., 2000).
Electro-oxidation of the organic extract at the higher oxidation potential produced a maximum concentration of syringaldehyde (0.080% ± 2.606 x 10-2%) and vanillin (0.22% ± 8.73 x 10-2%) within the first half hour (Fig. 13). Thereafter the concentration of both compounds decreased steadily.
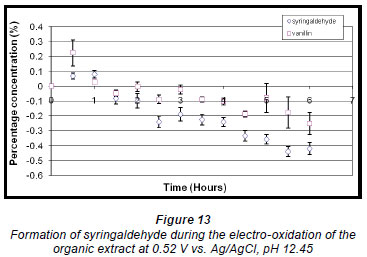
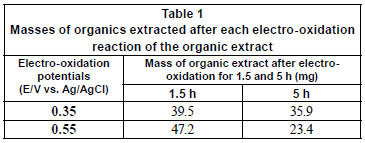
The mass of organics present after electro-oxidation was also determined and found to vary (Table 1). A significant reduction occurred, particularly during syntheses that were performed at higher oxidation potentials for the same period of time as syntheses performed at lower potentials, suggesting that electro-oxidation can also be used as a means of reducing the amounts of organics present in the effluent before it is pumped out to sea.
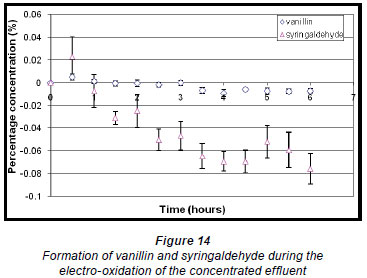
During the electro-oxidation of the concentrated form of the effluent, the percentage concentrations of vanillin and syringaldehyde both increased initially and reached a maximum of 0.0051% (± 0.2581 x 10-2%) and 0.023% (± 1.757 x 10-2 %), respectively, within the first half hour of the reaction (Fig. 14). Thereafter, a steady decrease in percentage concentration of syringaldehyde is noted, whereas the percentage concentration of vanillin decreased to a much lesser extent. This again shows that the compounds of interest would need to be extracted from the solution to prevent them from being destroyed.
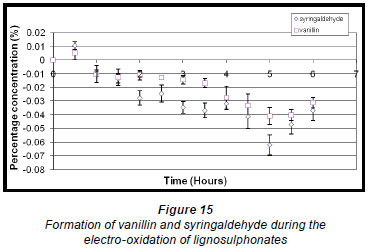
The maximum concentration of syringaldehyde (0.011% ± 0.274 x 10-2%) was reached within the first half hour of the electro-oxidation of lignosulphonates and thereafter decreased. Vanillin also reached a maximum within the first half hour but at a much lower percentage concentration (0.0051% ± 0.4715 x 10-2%), and thereafter its concentration also decreased (Fig. 15).
A literature survey of electro-oxidation reactions carried out on lignin showed the formation of vanillin from the electro-oxidation of alkaline Kraft lignin (Parpot et al., 2000). However the electro oxidation reactions had been carried out at 750 A/m2 whereas the electro-oxidation reactions in this work were performed at approximately 3 A/m2. In order to provide a comparison of the results obtained in this work with that published in literature, the percentage concentration of vanillin obtained was converted to millimolarity (mM) and multiplied 250 times to allow for a theoretical scale up of the current passing through the solution. A result of 0.127 mM was obtained based on the electro-oxidation of a concentrated form of the effluent made up to a solution of 0.15% (m/v) and 0.124 mM of vanillin formed based on the electro oxidation of 0.15% (m/v) lignosulphonate. These results, using lignin obtained from Saiccor's acid bisulphite-based pulping process, compares well with that obtained in literature, where a 0.07 mM concentration was obtained based on constant current electrolysis of a 0.5% alkaline Kraft lignin solution (Parpot et al., 2000). This is the first time that such investigations of lignin obtained from Sappi Saiccor's acid bisulphite pulping process have been reported. In addition, this work showed that a maximum amount of vanillin was obtained within half an hour of electro-oxidation, compared to 50 min reported in the literature for the electro oxidation of alkaline Kraft lignin. Thus, the results obtained for electro-oxidation reactions of lignin from Saiccor's effluent based on a theoretical scale-up show an increase in the amount of vanillin formed. No previous results on the amounts of syringaldehyde formed from electro-oxidation reactions of acid bisulphite pulping effluent could be found for comparison.
Conclusions
The electro-oxidation reactions of the pure compounds syringaldehyde and syringaresinol show that electro-oxidation reactions of lignin monomers and lignans are possible. Electrooxidation of syringaldehyde produces its corresponding acid as well as dimers. These dimers, although present in smaller quantities, provide evidence for the polymerisation of the lignin monomer, which is known to occur in reactions based on radical formation (Pearl, 1952; Pardini et al., 1991; Pearl, 1957; Altwicker, 1967). Electro-oxidation reactions of the extracted organic component of the effluent, a concentrated form of the effluent and a solution in water of lignosulphonate powder obtained from Lignotech, all showed the formation of syringaldehyde and a smaller amount of vanillin. The results obtained for vanillin compare favourably with that reported in literature; however, the results reported here for syringaldehyde are the first reported on the formation of syringaldehyde obtained from the electro-oxidation of acid bisulphite pulping effluent (Parpot et al., 2000). The compounds of interest must also be removed from the reaction mixture as they form to prevent them from being destroyed.
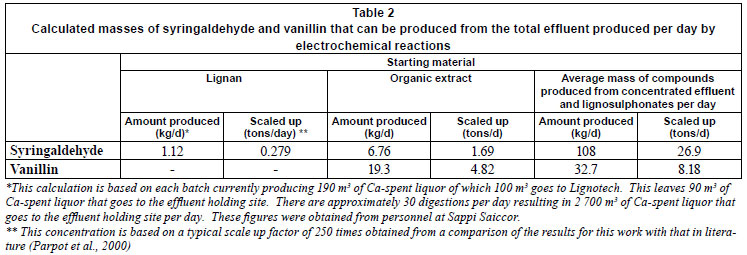
The higher concentrations produced at higher potentials were used to calculate the masses of vanillin and syringaldehyde that could possibly be produced after scaling up the reaction 250 times, taking into account the flow rates of the effluent to the effluent holding site. (A scale up of 250 times was obtained from a comparison of results for this work with that found in literature). Table 2 shows that large amounts of syringaldehyde and vanillin can be produced by scaling up the reaction concomitant with the large known volumes of 2 700 m3 of effluent produced per day, even though the actual percentage concentrations obtained in this study were low.
Higher temperatures should also be considered to further improve the yields of vanillin and syringaldehyde. Further pilot plant studies are needed to determine the industrial feasibility of this study and to optimise the conditions for maximum production of vanillin and syringaldehyde.
In addition, the various electro-oxidation reactions carried out on the effluent also show a reduction in the amount of vanillin and syringaldehyde over long periods of electrolysis. Thus, electrooxidation of the effluent can also be used as a means of reducing the organic content of the effluent before it is discharged to sea. The preliminary results obtained in this work show a significant reduction in the amounts of the major organics present in the effluent after long periods of electro-oxidation of the effluent, and so effective and correct use of this technique could result in a significant reduction of the major organic components present in the total amount of effluent discharged. Studies using electro-oxidation as a means of degrading dyestuffs in water have been performed and have shown significant reductions in colour compounds (Mphephu, 1996; Allen et al., 1995). Anodic oxidation reactions are also known to oxidise organic compounds to less harmful products and prolonged electro-oxidation can eventually destroy the compound (Mphephu, 1996; Allen et al., 1995; Simonsson, 1997; El-Ashtoukhy et al., 2009; Patel and Suresh, 2008; Ugurlu et al., 2008; Ma et al., 2007).
Electro-oxidation has been found to be an environmentally favourable process that does not produce toxic products and is a selective reaction, allowing for easy control in reducing the amount of organics present in the effluent. Overall, the electrooxidation of Sappi Saiccor's effluent provides a possible route for the formation of compounds of commercial value without the addition of further chemicals.
Acknowledgements
Funding from Sappi Saiccor via Mr Weightman is acknowledged, as is the National Research Foundation (NRF), South Africa. We are grateful to Mr John Thubron for effluent samples and pulping process information and to Mr Dilip Jagjivan for NMR spectra and Mr Bret Parel for assisting with GC-MS and LC-MS data collection.
References
ALLEN S, KHADER KYH and BINO M (1995) Electrooxidation of dyestuffs in waste waters. J. Chem. Tech. Biotechnol. 62 (2) 111-117. [ Links ]
ALTWICKER ER (1967) The chemistry of stable phenoxy radicals. Chem. Rev. 67 (5) 475-527. [ Links ]
ANNUNZIATINI C, BAIOCCO P, GERINI MF, LANZALUNGA O and SJOGREN B (2005) Aryl substituted N-hydroxyphthalimides as mediators in the laccase-catalysed oxidation of lignin model compounds and delignification of wood pulp. J. of Molec. Catal. B: Enzymatic. 32 (3) 89-96. [ Links ]
ARZOLA KG, AREVALO MC and FALCON MA (2009) Catalytic efficiency of natural and synthetic compounds used as laccasemediators in oxidising veratryl alcohol and a kraft lignin, estimated by electrochemical analysis. Electrochim. Acta 54 (9) 2621-2629. [ Links ]
BANGANI V (1998) Homoisoflavonoids and Stilbenoids from Scilla Species. Master of Science Dissertation, University of Natal, Durban. [ Links ]
CANIZARES P, DIAZ M, DOMNGUEZ JA, GARCA-GMEZ J and RODRIGO MA (2002) Electrochemical oxidation of aqueous phenol wastes on synthetic diamond thin-film electrodes. Ind. Eng. Chem. Res. 41 (17) 4187-4194. [ Links ]
DEMIN VA, BOGOMOLOV BD, KARMANOV AP and DAVYDOV VD (1980) Structural changes in sulfate lignin during electrochemical oxidation. Koksnes Kimija 4 82. URL: https://scifinder.cas.org (Accessed 22 December 2008). [ Links ]
EL-ASHTOUKHY E-SZ, AMIN NK and ABEDLWAHAB O (2009) Treatment of paper mill effluents in a batch-stirred electrochemical tank reactor. Chem. Eng. J. 146 205-210. [ Links ]
ELVING PJ and KRIVIS AF (1958) Voltammetric studies with graphite indicating electrode. Anal. Chem. 30 (10) 1645-1648. [ Links ]
GAYLOR VF, CONRAD AL and LANDERL JH (1957) Use of wax-impregnated graphite electrode in polarography. Anal. Chem. 29 (2) 224-228. [ Links ]
GAYLOR VF, ELVING PJ and CONRAD AL (1953) Polarographic oxidation of phenolic compounds. Anal. Chem. 25 (7) 1078-1082. [ Links ]
HEDENBERG JF and FREISER H (1953) Anodic voltammetry of phenols. Anal. Chem. 25 (9) 1355-1358. [ Links ]
INIESTA J, MICHAUD PA, PANIZZA M, CERISOLA G, ALDAZ A and COMNINELLIS CH (2001) Electrochemical oxidation of phenol at boron-doped diamond electrodes. Electrochim. Acta 46 3573-3578. [ Links ]
JAMES JH (1899) An electrolytic study of benzoin and benzyl. J. Am. Chem. Soc. 21 (10) 889-910. [ Links ]
KOVALENKO EI, SHIFRON EM and RASKIN MN (1973) Electrochemical oxidation of the natural polymer lignin. Novosti Elektrokhim. Org. Soedin., Tezisy Dokl. Vses. Soveshch. Elektrokhim. Org. Soedin. URL: https://scifinder.cas.org (Accessed 22 December 2008). [ Links ]
KOVALENKO EI, SHIFRON EM and RASKIN MN (1976) Electrochemical oxidation of the natural polymer lignin. Trudy Novocherkasskogo Politekhnicheskogo Instituta imeni Sergo Ordzhonikidze 320 69. URL: https://scifinder.cas.org (22 December 2008). [ Links ]
KOVALENKO EI, SMIRNOV VA and SHALIMOV VN (1977) Effect of anode material and electrolysis time on the directivity of the electrochemical oxidation of hydrolytic lignin . Zhurnal Prikladnoi Khimii (Sankt-Peterburg, Russian Federation) 50 1741. URL: https://scifinder.cas.org (22 December 2008). [ Links ]
KOVALENKO EI, POPOVA OV, ALEKSANDROV AA and GALIK-YAN TG (2000) Electrochemical modification of lignin. Russ. J. Electrochem. 36 (7) 706-711. [ Links ]
LAW HD (1906) Electrolytic oxidation. J Soc. Chem. Trans. 89 1437-1453. [ Links ]
LUND H (1957) Electroorganic preparations IV. Oxidation of aromatic hydrocarbons. Acta Chem. Scand. 11 (8) 1323-1330. [ Links ]
MA H, WANG B and WANG Y (2007) Application of molybdenum and phosphate modified kaolin in electrochemical treatment in paper mill wastewater. J. Hazard. Mater. 145 417-423. [ Links ]
MAJUMDER PL and PAL S (1993) Cumulatin and Tristin, Two bibenzyl derivatives from the orchids Dendrobium cumulatum and Bulbophyllum triste. Phytochem. 32 (6) 1561-1565. [ Links ]
MOODLEY B, MULHOLLAND DA and MARSH JJ (2003) The characterisation of organic components in the calcium and magnesium effluent streams at Sappi Saiccor. Water SA 29 (3) 237-240. [ Links ]
MORRIS JB and SCHEMPF JM (1959) Voltammetric studies at the graphite electrode in quiet solutions. Anal. Chem. 31 (2) 286-291. [ Links ]
MPHEPHU NA (1996) An Electrochemical Study of Azo Dyes. Master of Science Dissertation, University of Natal, Durban. [ Links ]
PARDINI VL, SMITH CZ, UTLEY JHP, VARGAS RR and VIERT-LER H (1991) Electroorganic reactions. 38. Mechanisms of electrooxidative cleavage of lignin model dimers. J. Org. Chem. 56 (26) 7305-7313. [ Links ]
PARPOT P, BETTENCOURT AP, CARVALHO AM and BELGSIR EM (2000) Biomass conversion: attempted electrooxidation of lignin for vanillin production. J. Appl. Electrochem. 30 727-731. [ Links ]
PATEL UD and SURESH S (2008) Electrochemical treatment of pentachlorophenol in water and pulp bleaching effluent. Sep. Purif. Technol. 61 115-122. [ Links ]
PEARL IA (1952) Reactions of vanillin and its derived compounds. XVI. The synthesis of vanillil. J. Am. Chem. Soc. 74 (17) 4260-4262. [ Links ]
PEARL IA (1957) Reactions of vanillin and its derived compounds XXVII. Synthesis in the syringyl series. J. Org. Chem. 22 (10) 1229-1232. [ Links ]
SIMONSSON D (1997) Electrochemistry for a cleaner environment. Chem. Soc. Rev. 26 181-189. [ Links ]
SMITH C and UTLEY JHP (1989) Biomass electrochemistry: anodic oxidation of an organo-solv lignin in the presence of nitroaromatics. J. Appl. Electrochem. 19 535-539. [ Links ]
TIAN M, WEN J, MACDONALD D, AMUSSEN RM and CHEN A (2010) A novel approach for lignin modification and degradation. Electrochem. Commun. 12 527-530. [ Links ]
UGURLU M, GURSES A, DOGAR C and YALCIN M (2008) The removal of lignin and phenol from paper mill effluents by electrocoagulation. J. Environ. Manage. 87 420-428. [ Links ]
UTLEY JHP and SMITH CZ (1986) Electrochemical treatment of lignins. WO Patent No. 87/03014. [ Links ]
UTLEY JHP and SMITH CZ (1987) Electrochemical treatment of lignins. EP Patent No. 0 245 418. [ Links ]
UTLEY JHP and SMITH CZ (1988) Electrochemical treatment of lignins. US Patent No. 4 786 382. [ Links ]
VAGIN MU, TRASHIN SA and KARYAKIN AA (2006) Corrosion protection of steel by electropolymerized lignin. Electrochem. Commun. 8 60-64. [ Links ]
VAREY P (ed.) (1987) Lignin the feedstock of the future? The Chem. Eng. 438 26. [ Links ]
VERMILLION FJ (Jr) (1963) Anodic Reactions of Simple Phenolic Compounds. Doctoral Dissertation, The Institute of Paper Chemistry, Wisconsin. [ Links ]
YOSHIDA K (1984) Electro-oxidation in Organic Chemistry: The Role of Cation Radicals as Synthetic Intermediates. John Wiley and Sons, New York. [ Links ]
Received 28 October 2009; accepted in revised form 8 November 2010.
* To whom all correspondence should be addressed. +2731 2602796; fax: +2731 2603091; e-mail: moodleyb3@ukzn.ac.za


{kind=link}