










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkWater SA
versão On-line ISSN 1816-7950
versão impressa ISSN 0378-4738
Water SA vol.36 no.5 Pretoria Out. 2010
REVIEW
The fundamental mechanism of aqueous contaminant removal by metallic iron
C Noubactep*
Angewandte Geologie, Universität Göttingen, Goldschmidtstraße 3, D - 37077 Göttingen, Germany
ABSTRACT
Contaminant co-precipitation with continuously generated and transformed iron corrosion products has received relatively little attention in comparison to other possible removal mechanisms (adsorption, oxidation, precipitation) in Fe0/H2O systems at near neutral pH values. A primary reason for this is that the use of elemental iron (Fe0) in environmental remediation is based on the thermodynamic-founded premise that reducible contaminants are potentially reduced while Fe0 is oxidised. However, co-precipitation portends to be of fundamental importance for the process of contaminant removal in Fe0/H2O systems, as the successful removal of bacteria, viruses and non reducible organic (e.g. methylene blue, triazoles) and inorganic (e.g. Zn) compounds has been reported. This later consideration has led to a search for the reasons why the importance of co-precipitation has almost been overlooked for more than a decade. Three major reasons have been identified: the improper consideration of the huge literature of iron corrosion by pioneer works, yielding to propagation of misconceptions in the iron technology literature; the improper consideration of available results from other branches of environmental science (e.g. CO2 corrosion, electrocoagulation using Fe0 electrodes, Fe or Mn geochemistry); and the use of inappropriate experimental procedures (in particular, mixing operations). The present paper demonstrates that contaminant co-precipitation with iron corrosion products is the fundamental mechanism of contaminant removal in Fe0/H2O systems. Therefore, the 'iron technology' as a whole is to be revisited as the 'know-why' of contaminant removal is yet to be properly addressed.
Keywords: adsorption, co-precipitation, iron corrosion, iron oxhydroxides, zerovalent iron
INTRODUCTION
The use of iron-based alloys (elemental iron, zerovalent iron, Fe0 materials or simply Fe0) for environmental remediation is certainly one of the ideas that have fascinated environmental scientists during the last 2 decades (Gillham and O'Hannesin, 1994; Lipczynska-Kochany et al., 1994; Matheson and Tratnyek, 1994; Schreier and Reinhard, 1994; O'Hannesin and Gillham, 1998; Bigg and Judd, 2000; Scherer et al., 2000; Lee et al., 2004; Bojic et al., 2007; Henderson and Demond, 2007; Laine and Cheng, 2007; Cundy et al., 2008; Jeen et al., 2008; Thiruvenkatachari et al., 2008). After the successful implementation of the first iron walls (O'Hannesin and Gillham, 1998), and the controversial use of iron nano-particles for quantitative contaminant reduction at source zones (Gillham, 2003, Noubactep and Care, 2010a), Fe0 materials could be regarded as the media of choice for all contamination events. Presently, around 120 Fe barriers are installed worldwide and most of them currently meet design specifications (Jambor et al., 2005; Henderson and Demond, 2007; Cundy et al., 2008).
The primary use of Fe0 materials in environmental remediation is based on the premise that quantitative contaminant removal in Fe0/H2O systems is mostly due to contaminant reduction through electrons from the metal body (Matheson and Tratnyek, 1994; Weber, 1996, O'Hannesin and Gillham, 1998). Because of the electrochemical nature of aqueous corrosion, this is only possible:
• At the Fe0 surface
• Through defects in the metal such as pits
• At the surface of an overlaying electrically conductive oxide-film.
However, given that under typical ranges of sub-surface pH (6 < pH < 9) (e.g. Aleksanyan et al., 2007) there are several other possible contaminant removal mechanisms (adsorption, co-precipitation, indirect reduction) in Fe0/H2O systems (e.g. Stipp et al., 2002), it is doubtful whether direct reduction may be the major contaminant removal pathway as it is commonly assumed.
The concept of reductive transformation (degradation of organics and precipitation of inorganics) in Fe0/H2O systems persists in current literature, despite a number of papers that have attempted to theoretically and experimentally point out their inherent shortcomings, as summarised by Noubactep (2007; 2008; 2010). In particular, the observed lag time between Fe0 supply and quantitative contaminant removal is a strong argument against direct reductive transformations (electrons from Fe0). Evidently, the cited papers (Noubactep, 2007; Noubactep, 2008; Noubactep, 2010; Noubactep et al., 2010a) and related papers (Noubactep, 2009a-g; Noubactep and Schoner, 2009; Noubactep et al., 2009a; 2009b; Noubactep, 2010b; Noubactep and Care, 2010a; Noubactep and Schoner, 2010a) are not clear enough in their explanation to convince many authors of current articles dealing with contaminant removal in Fe0/H2O systems (Elsner et al., 2007; Jeen et al., 2008; Katsoyiannis et al., 2008; Wang and Cheng, 2008; Kang and Choi, 2009; Tratnyek and Salter, 2010). An understanding of how aqueous contaminants are effectively removed in the presence of Fe0 is essential for proper designing of the reactive barrier (e.g. used amounts of Fe0 or/and barrier thickness) and discussing the stability of removed contaminants.
The results of previous works aimed at questioning the validity of the reductive transformation concept can be summarised in 3 major points:
• The dynamic nature of the process of corrosion product generation has not been considered (Noubactep, 2007; Noubactep 2010a; Noubactep et al., 2010a)
• The combined problem of mass transport and chemical reaction in Fe0/H2O systems has not been properly considered (Noubactep 2008, Noubactep, 2009c; Noubactep, 2009d)
• The results available from other branches of science using Fe0 (aqueous Fe0 corrosion, electrocoagulation using iron electrodes, hydrometallurgy, Fe0 in the petroleum industry) have not been properly considered (Noubactep and Schoner, 2009; Noubactep, 2010b; Noubactep and Schoner, 2010a).
As a result adsorption (onto) and co-precipitation (with) iron corrosion products as fundamental contaminant removal mechanism has been suggested (Noubactep, 2007; 2008; 2010a) and experimentally validated (Noubactep et al., 2003; Noubactep et al., 2006a; Noubactep, 2009c; Noubactep 2009e; Noubactep et al., 2009a; Ghauch et al., 2010a; Ghauch et al., 2010b).
This paper attempts to further clarify the fundamental mechanism of contaminant removal in Fe0/H2O systems at near neutral pH values by approaching the subject from a new perspective: the similitude between Fe0/H2O systems and geochemical systems involving iron and manganese precipitation is underlined. As recalled above, the concept of contaminant reductive transformation through Fe0 is the main prop that holds up the iron reactive barrier technology. The origin of the instable premise is identified, an alternative and more reliable interpretation for the process of contaminant removal in Fe0/ H2O systems is proposed, and recommendations for future works are made.
The process of aqueous Fe0 oxidation
Elemental iron (Fe0) is unstable under environmental conditions (Knowlton, 1928; Brondel et al., 1994; Aleksanyan et al., 2007; Nesic, 2007). Its oxidation yields FeII species the stability of which is mostly dependent on the pH value and the redox potential (Liu and Millero, 1999). In the pH range relevant for natural waters, FeII species are of limited stability and are very sensitive to molecular oxygen (O2) (e.g. Aleksanyan et al., 2007; Nesic, 2007). The oxidation of FeII species by molecular oxygen is quite rapid. Resulting FeIII species (primarily Fe3 + ions) readily hydrolyse, precipitate (Fig. 1) and transform to oxides ([([Fe(H2O)6]3+)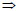 Fe(OH)3 (aq)Fe(OH)3 (s)oxides. Depending on the O2 availability, various oxides (FeIIO, FeIIFeIIIO4, FeIIIOOH, FeIII2O3) are generated (Mackenzie et al., 1999; Wilkin et al., 2003; Mielczarski et al., 2005). The theoretical ratio between the volume of corrosion products and the volume of iron in the crystal structure varies between 2.0 for Fe3O4 and 6.40 for Fe(OH)3.3H2O (Care et al., 2008).
Fe(OH)3 (aq)Fe(OH)3 (s)oxides. Depending on the O2 availability, various oxides (FeIIO, FeIIFeIIIO4, FeIIIOOH, FeIII2O3) are generated (Mackenzie et al., 1999; Wilkin et al., 2003; Mielczarski et al., 2005). The theoretical ratio between the volume of corrosion products and the volume of iron in the crystal structure varies between 2.0 for Fe3O4 and 6.40 for Fe(OH)3.3H2O (Care et al., 2008).
The process of abiotic and biotic FeII oxidation is widely described in the geochemical literature (Davison and Seed, 1983; Millero et al., 1995; Morgan and Lahav, 2007). The discussion of the involved processes is often coupled to those occurring during abiotic and biotic Mn2+ oxidation (Postma, 1985; Post, 1999; Martin, 2005 and references therein). One of the central features of these systems is the fact that Fe2+/ Mn2+ oxidative precipitation at near neutral pH values is accompanied by inorganic and organic molecule entrapment (co-precipitation). The process of co-precipitated contaminant release during reductive dissolution of Fe and Mn oxides is almost an independent branch of geochemical research (Stone and Morgan, 1984a; Stone and Morgan, 1984b; Stone, 1987a; Stone, 1987b; Weaver and Hochella 2003; Shi and Stone, 2009a; Shi and Stone, 2009b). This fact is mostly materialised by an independent step in schemes used for sequential extraction to reductively dissolve Fe and Mn hydroxides (using mostly NH2OH HCl as reducing agent) to liberate sequestrated metal species (Tessier et al., 1979; Kasalainen and Yli-Halla, 2003; Madrid et al., 2007). Therefore, it is rather surprising that contaminant co-precipitation (the process and its importance) with continuously generated and ageing corrosion products has almost been overlooked in the 'iron technology' literature for more than a decade. Two possible reasons for this are:
• The extreme specialisation in modern scientific research. This specialisation creates new branches and researchers starting at a later time scale may loss the contact to the mother branch.
• The educational background of individual researchers as environmental scientists is of very heterogeneous origin. It is not likely that the subtlety and scientific diversity of the physical and chemical processes involved in iron corrosion will be easily understood by individual scientists.
Readers interested in more detail on the process of co-precipitation are encouraged to read 2 excellent works by Crawford et al. (1993a, 1993b). It should be noted that:
• Co-precipitation is a primarily unspecific removal mechanism that occurs whenever an abundant species (iron) precipitates (here as oxide) in the presence of trace amounts of foreign species (here contaminants)
• Co-precipitated contaminants are not likely to be released into the environment unless iron oxides are dissolved (Stipp et al., 2002; Noubactep et al., 2003; Noubactep et al., 2006a; Noubactep et al., 2006b; Noubactep, 2009c; Noubactep et al., 2009a; Ghauch et al., 2010a).
Therefore, although contaminants are not necessarily reduced in Fe0/H2O systems, they are successfully removed and strongly sequestrated in the matrix of iron oxides. This is the major reason why no carbon balances between reactants and products have ever been successfully done for many chlorinated hydrocarbons (Lee et al., 2004). The reported lack of mass balance indicates that reduction as removal mechanism by Fe0 is not certain. However, whether reaction products are environmentally safe or not, the concept of contaminant co-precipitation with iron corrosion products suggests that contaminants, their possible reaction products, and all possible foreign species, are sequestrated in the matrix of iron oxides and are stable so far as oxides are not dissolved. This conclusion is supported by the observation that even bacteria (Hussam and Munir, 2007; Diao and Yao, 2009) and viruses (You et al., 2005) are successfully removed in Fe0/H2O systems. The latter observation has led to the suggestion of metallic iron as a universal filter material in small above-ground walls and household filters for treatment of waters of unknown quality (Noubactep and Woafo, 2008; Noubactep et al., 2009b; Noubactep and Schoner, 2010b). If successfully developed, this simple idea could enable universal access to safe drinking water in remote areas around the world. It should be kept in mind that underground reactive barriers are primarily implemented at sites with contaminant concentrations that are not far above the MCL (maximum contaminant level). Thus, the total mass of contaminant passing through the barrier over its entire lifetime is small relative to the available Fe0 within the barrier (Palmer and Wittbrodt, 1991). Given the diversity of contaminant removal agents (Fe0 and corrosion products) and processes (adsorption, co-precipitation, reduction), the question to be answered is how long Fe0 consumption (or corrosion) could/will enable satisfactory contaminant removal.
The discussion above shows clearly that pioneer works on contaminant removal in Fe0/H2O systems overlooked an important pathway of contaminant removal. Several other aspects have not been properly considered. For example, since iron corrosion continues despite oxide-film formation, available information from other branches of science using Fe0 materials should have been exploited as thought-provoking impulses for a better comprehension of the processes of contaminant removal in Fe0/H2O systems (Noubactep and Schoner, 2009; Noubactep and Schoner, 2010a). An excellent example is a mechanistic model for carbon dioxide corrosion of mild steel in the presence of protective iron carbonate films, proposed by Nesic and his co-authors (Nesic and Lee, 2003; Nesic et al., 2003; Nordsveen et al., 2003). These 3 papers have discussed very useful numerical simulation results, which are yet to be 'discovered' by 'iron technology' researchers.
For the sake of clarity the next section will present the process of contaminant co-precipitation in some detail and show how it has been considered in the field of 'iron technology' to date.
Contaminant co-precipitation
Co-precipitation of substances with metal (mainly Fe, Al, Mn) oxides (Crawford et al., 1993a; 1993b; Karthikeyan et al., 1997) occurs in natural environments and is a remediation technology used to remove toxic elements (mainly metal, metalloid and radionuclides) from drinking water (Scott et al., 1995), wastewater (Rangsivek and Jekel, 2005) and hydrometallurgical solutions (Dutrizac, 1991).
Co-precipitation is a process in which normally soluble species are carried out of solution by sequestration in a precipitating phase. In other words, co-precipitation is the simultaneous removal of a foreign species (including living bacteria and viruses) during the formation of a primary metal precipitate (mainly Fe and Mn hydroxides). Co-precipitation can entail:
• Contaminant adsorption onto freshly formed hydrous oxide colloids (surface adsorption)
• Solid solution formation by contaminant incorporation into the hydrous oxide lattice (mixed-crystal formation)
• Mechanical enclosure of contaminant-containing solution by the precipitate (occlusion or mechanical entrapment) (Crawford et al., 1993a; 1993b; Karthikeyan et al., 1997).
Co-precipitation in any real-world system is necessarily a combination of these 3 processes. It is important to notice that only mixed-crystal formation is primarily specific for inorganic compounds as the atomic radii have to be comparable.
Knowledge of the co-precipitation of dissolved organic matter with iron hydroxides is still limited (Satoh et al., 2006 and references therein). The mechanism of organic contaminant co-precipitation in Fe0/H2O systems can be described as follows: FeII species from Fe0 oxidation are released in the aqueous solution and are further oxidised to FeIII species by dissolved O2 or other oxidants (e.g. MnO2). The solubility of FeIIIcompounds at pH > 5.0 is very small; therefore, precipitates are formed (Liu and Millero, 1999; Rickard, 2006; Aleksanyan et al., 2007 - see Fig. 1). The most common FeIII species in natural waters is hydrated FeIII hydroxide oxide, which is commonly positively charged (Satoh et al., 2006). The FeIII precipitates electrostatically co-precipitate negatively charged colloids and dissolved substances, including organic matter (Sugiyama and Kumagai, 2001; Schwertmann et al., 2004; Satoh et al., 2006, Violante et al., 2007).
Although a Fe0/H2O system is rigorously a domain of precipitating iron oxide, research on using Fe0 for environmental remediation over the past 20 years has been based on the fortuitous observation that trichloroethylene is removed from aqueous solution in the presence of metallic iron (Reynolds et al., 1990). Therefore, Fe0 materials have been mostly tested and used for the removal of reducible substances (chlorinated hydrocarbons, nitro aromatics, CrVI, UVI). However, adsorption and co-precipitation have been widely and independently described as 2 active mechanisms to trap contaminants from contaminated waters (e.g. Langmuir, 1997; Stipp et al., 2002; Schwertmann et al., 2004). Fixation of contaminants in solid phases can remove ions from solution retarding their transport. When a contaminant is incorporated in the matrix rather than simply adsorbed at the surface, it is less available and can be considered as 'immobilised' in the environment, at least until the host phase is dissolved (Noubactep et al., 2006b). Therefore, contaminant co-precipitation is a suitable mechanism for long-term remediation under conditions of low solubility of the host phase (pH > 4 for iron hydroxides). This has been acknowledged and precipitation walls, using for example lime and calcium carbonate, have been engineered for the remediation of heavy metal contaminated groundwater (Komnitsas et al., 2004; Lee et al., 2007; Sibrell et al., 2007).
Concerning Fe0 reactive barriers, besides reduction by Fe0 (and FeII), contaminant adsorption at hydrous oxide surfaces at near neutral pH values (5 < pH < 9) is widely reported in the literature (Matheson and Tratnyek, 1994; Mantha et al., 2001; Stipp et al., 2002; Henderson and Demond, 2007), with sporadic mention of co-precipitation almost exclusively for inorganics (Noubactep et al., 2003; Lien and Wilkin, 2005; Rangsivek and Jekel, 2005). On the contrary, inorganic contaminant co-precipitation in Fe0/H2O systems at pH < 5 (mostly acid mine drainage) has been largely reported (Wilkin and McNeil, 2003; Bartzas et al., 2006; Komnitsas et al., 2007) and will not be further considered in this work. However, according to the definition of co-precipitation recalled above, regardless of the pH value, a Fe0/H2O system should be regarded as the domain of co-precipitating iron hydroxide. Each species (living, organic, inorganic) flowing into this domain should be regarded as foreign species that will co-precipitate with iron hydroxide (Noubactep, 2009f). Taken alone, this stoichiometric argument is a very strong argument for co-precipitation as fundamental mechanism of contaminant removal by Fe0. Moreover, contaminants and their derivates are co-precipitated and are not likely to be quantitatively released to the environment. Furthermore, even though some removed contaminants may be released while iron oxide is structurally transformed (Fe(OH)2/Fe(OH)3 FeOOH, Fe2O3, Fe3O4), they will be entrapped within the reactive walls by continuously generated new corrosion products. This is very likely the reason why, despite obvious reductive reactivity loss by Fe0 due to surface coverage by corrosion products, installed reactive Fe0 walls currently meet design specifications.
The next section gives an overview of the current considerations of the mechanism of contaminant removal in Fe0/ H2O systems, and an alternative taking co-precipitation into account.
Common considerations and their limitations
In investigating the removal of many chlorinated hydrocarbons in Fe0/H2O systems, no carbon balances between reactants and products have ever been successfully done (Lee et al., 2004). The situation is similar for inorganic species. Although reductive dissolution of Fe oxides is a well-known procedure to liberate bounded species (sequential extraction as discussed above), very few studies have used reductive dissolution of corrosion products for mass balance (Noubactep et al., 2006b). Furthermore, complexing solutions like Na2CO3 or NaHCO3 for uranium have been used for speciation (Gu et al., 1998). However, uranium species (both UVI and UIV) are soluble in carbonate solutions while iron species (FeII and FeIII) are not. Therefore, co-precipitated uranium will not be released in carbonate solutions. Co-precipitated uranium is regarded by these investigators as reductively precipitated by Fe0.
Contaminant removal in Fe0/H2O systems has been assumed to be driven by an electrochemical reduction between redoxsensitive species and elemental iron (Laine and Cheng, 2007; Matheson and Tratnyek, 1994; Powell et al., 1995; Schreier and Reinhard, 1994; Weber, 1996). These considerations suggest that all contaminants with an electrode potential higher than the electrode potential of the redox couple FeII/Fe0 (-0.44 V) should be reduced in Fe0/H2O systems. However, the redox potential (E0) is a thermodynamic parameter. As such, E0-values only indicate the possibility of a reaction. Therefore, the reaction rate and the reaction mechanism should have been properly demonstrated for individual compounds. This has not been the case.
In discussing the mechanism of contaminant removal in Fe0/H2O systems, a general consensus is that elemental iron serves as reducing agent (direct reduction through Fe0 - O'Hannesin and Gillham 1998) and the overlaying oxide film serves as catalyst (e.g. indirect reduction through FeII). This consensus totally neglects the fact that oxide film formation is a dynamic process, in the course of which contaminants are certainly entrapped in the mass of precipitating iron oxides (co-precipitation). Moreover, the oxide-film should be regarded as a mixture of oxides of different reactivity, acting both as adsorbents and coprecipitants. Adsorbed and co-precipitated contaminants can be subsequently reduced by direct or/and indirect reduction. Therefore, indirect reduction is more likely than direct reduction as the presence of a conductive oxide-film is necessary for direct reduction (Noubactep, 2007; Noubactep, 2008). Note that, despite the 'broad consensus' on reduction degradation of organics (O'Hannesin and Gillham 1998), some investigators could not traceably identify the role of Fe0 in contaminant reduction in Fe0/H2O systems. (Lavine et al., 2001; Lee et al., 2004; Jiao et al., 2009). For example, Jiao et al. (2009) found that the presence of carbon tetrachloride (CT) significantly accelerated aqueous iron corrosion and the iron corrosion was helpful for the reductive dechlorination of CT. CT was already investigated in the seminal work of Matheson and Tratnyek (1994) as model contaminant and further investigated over the years by several research groups univocally reporting on reductive dechlorination by Fe0 (e.g. Helland et al. 1995; Erbs et al., 1999; Li and Farrell, 2000; Li and Farrell, 2001; Doong et al., 2003; Meng et al., 2006). Jiao and colleagues (2009) concluded that the removal efficiency of CT is strongly dependent on the extent of iron corrosion in aqueous solutions at different pH values (0.4 < pH < 14). Clearly, the results of Jiao et al. (2009) demonstrate that CT reduction is not a direct reduction by Fe0, validating the adsorption/co-precipitation concept (Noubactep, 2007; Noubactep, 2008).
Three possible functions have been attributed to the universal oxide-film on Fe0 (Scherer et al., 2000; Huang and Sorial, 2007):
• The oxide film serves as a physical barrier inhibiting electron transfer from Fe0 to the electron acceptor contaminant and the electron transfer may occur from the metal to the dissolved substance through defects in the metal such as pits
• The oxide film mediates electron transfer from Fe0 to the contaminant by acting as a semiconductor
• The oxide film acts as a coordinating surface containing sites of FeII, which complexes with the contaminant and then reduces it.
It is evident, that this well-accepted model from Scherer et al. (2000) aims at explaining why contaminant reduction may occur despite the presence of the oxide film. Again the fact that oxide films continuously entrap adsorbed contaminants and contain already entrapped contaminants is not noticed.
One of the primary interests in the behaviour of contaminants in Fe0/H2O is in the manner in which they are removed from the aqueous phase. Adsorption on particles (Fe0, oxide film) is obviously the primary contaminant removal mechanism from the aqueous solution. Consequently, a primary consideration in predicting the behaviour of any contaminant should be to properly consider its affinity for available surfaces. Relatively few studies have properly incorporated the affinity between contaminants and solid surfaces in the discussion of experimental results. The situation is exacerbated by the use of inappropriate experimental conditions (Noubactep, 2007; Noubactep, 2009c; Noubactep, 2009e; Noubactep et al., 2009a). The affinity between contaminants and solid surfaces is mostly determined by: the solution pH, the contaminant characteristics (pKa-, pKs-values), and the point of zero charge (pHpzc) of solid surfaces (Nesic, 2007).
Alternative perspective
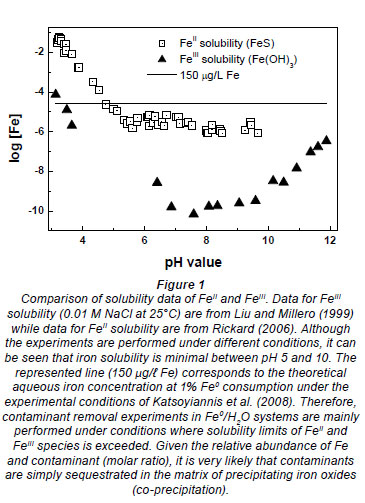
Aqueous contaminant removal in Fe0/H2O systems can be viewed as a 'trickle down' (Keeney-Kennicutt and Morse, 1985), in which a fraction of the targeted contaminant is continuously adsorbed onto in situ generated high adsorptive corrosion products. Contaminants are subsequently entrapped into the structure of ageing corrosion products (co-precipitation). In this situation, an equilibrium is not possible before Fe0 is depleted. Therefore, the use of adsorption isotherms (e.g. Freundlich, Langmuir) to interpret data of contaminant removal from short-term batch experiments is not justified. Furthermore, adsorbed or co-precipitated contaminants can be further reduced both by a direct and an indirect mechanism. The direct contaminant reduction is not always the most favourable reduction pathway (Noubactep, 2008; Noubactep, 2010a; Noubactep et al., 2010a). Contrary to this obviously plausible interpretation, it has been contended, without demonstration nor verification, that progressive contaminant removal in Fe0/H2O systems is the result of a direct reduction reaction according to Eq. (1). Equation (2) represents a reaction path that is possibly more favourable, not only thermodynamically (Noubactep, 2008; Noubactep, 2010a).

Where Ox and Red are the oxidised and the reduced form of the contaminant, respectively.
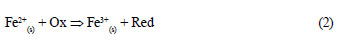
Fe2+(s) stands for surface adsorbed FeII or structural FeII (White and Peterson, 1996).
Concluding remarks
In investigating the process of contaminant removal in Fe0/ H2O systems, care is often taken to ensure that experimental data actually measure Fe0 surface oxidative dissolution reactions and not the process of product diffusion. Fe0 oxidative dissolution is supposedly coupled to contaminant reductive removal (Matheson and Tratneyk, 1994; Powell et al., 1995; Weber, 1996; Laine and Cheng, 2007). Traditionally, this objective is accomplished by mixing (shaking, stirring) the experimental vessels such that diffusion is faster than Fe0-surface chemistry. Unfortunately, any mixing operation inevitably accelerates the process of iron corrosion as the transport of primary corrosion products (FeII species) is accelerated (Polasek, 2007; Noubactep, 2009c; Noubactep, 2009g; Noubactep et al., 2009a). Transporting FeII ions away from the vicinity of the Fe0 surface inevitably disturbs (delays or even avoids) the process of oxide film formation. The discussion above has demonstrated the importance of oxide films for the process of contaminant removal. Therefore, besides the focus on the thermodynamic of the redox couple FeII/Fe0, mixing is probably the disturbing factor that has impeded or delayed the acceptance of the importance of adsorption and co-precipitation as basic contaminant removal mechanisms (Lee, et al., 2004; Noubactep, 2007; Noubactep, 2008; Noubactep, 2010a). In fact, the term mixing is confusing (Polasek, 2007) because it is randomly interchangeable, used to describe: transport mechanisms for reagent dispersion and homogenisation with water mixing (flash mixing), and agitation (flocculation mixing). However, each of these mechanisms (flash mixing and flocculation mixing) requires different flow characteristics in order to take place with maximum efficiency. For Fe0/H2O systems, depending on the mixing type and the mixing intensity, Fe0 and corrosion products may remain at the bottom of the reaction vessels or be held suspended in the aqueous phase. To be pertinent for real-world applications however, oxide-films have to be generated at the Fe0 surface.
Next to the disturbing effect on the oxide film formation, mixing has other obvious disadvantages for mechanistic investigations:
• Producing more corrosion products which are well known as good adsorbents for both organics and inorganics (Brown et al., 1999; Stipp et al., 2002), besides serving as catalyst for reduction through structural FeII (Riba et al, 2008 and references therein)
• Keeping corrosion products suspended and therefore facilitating the accessibility of the Fe0 surface for contaminants (Polasek, 2007).
Clearly, for the investigation of processes in Fe0/H2O systems, mixing should not be considered as a helpful tool to facilitate contaminant transport from the bulk solution to the Fe0 surface but rather as a possible disturbing factor (Lee et al., 2004; Noubactep, 2010a; Noubactep, 2009c; Noubactep et al., 2009a). This hypothesis could be verified experimentally while investigating the process of methylene blue discoloration by Fe0. It was shown that shaking intensities >50 min-1 are disturbing (Noubactep, 2009c). Consequently, in experiments investigating the process of contaminant removal by elemental iron materials, mixing operations (type and intensity) should be proven non-disturbing before being employed (Noubactep, 2007; Noubactep, 2009g; Noubactep et al., 2009a). Since in-situ formed corrosion products have to develop and remain on the Fe0 surface to mimic natural situations in subsurface reactive walls, mixing operations involving Fe0 particle movement (e.g. end-over-end agitation) inevitably introduced experimental biases. Depending on the used Fe0 particle sizes, all other mixing operations will more or less disturb the process of oxide film formation on Fe0 (Lee et al., 2004; Noubactep, 2010a).
For the further development of the 'iron technology', comprehensive investigations under more relevant experimental conditions are needed. Ideally a unified experimental procedure should be developed to enable better inter-laboratory comparison of results. In up-scaling results from well-thought-through experiments for the design of reactive barriers (Noubactep and Care, 2010b; Noubactep and Care, 2010c), available models for co-precipitation phenomena will be very useful (e.g. Komnitsas et al., 2006; Tokoro et al., 2008).
Acknowledgments
The manuscript was improved by the insightful comments from Angelika Schöner (FSU Jena). The work was supported by the Deutsche Forschungsgemeinschaft (DFG-No 626/2-2).
References
ALEKSANYAN AY, PODOBAEV AN and REFORMATSKAYA II (2007) Steady-state anodic dissolution of iron in neutral and closeto-neutral media. Protect. Met. 43 66-69. [ Links ]
BARTZAS G, KOMNITSAS K and PASPALIARIS I (2006) Laboratory evaluation of Fe0 barriers to treat acidic leachates. Miner. Eng. 19 505-514. [ Links ]
BIGG T and JUDD SJ (2000) Zero-valent iron for water treatment. Environ. Technol. 21 661-670. [ Links ]
BOJIC AL, PURENOVIC M, BOJIC D and ANDJELKOVIC T (2007) Dehalogenation of trihalomethanes by a micro-alloyed aluminium composite under flow conditions. Water SA 33 297-304. [ Links ]
BRONDEL D, EDWARDS R, HAYMAN A, HILL D, MEHTA S and SEMERAD T (1994) Corrosion in the oil industry. Oilfield Rev. 6 4-18. [ Links ]
BROWN JR GE, HENRICH VE, CASEY WH, CLARK DL, EGGLESTON C, FELMY A, GOODMAN DW, GRATZEL M, MACIEL G, MCCARTHY MI, NEALSON KH, SVERJENSKY DA, TONEY MF and ZACHARA JM (1999) Metal oxide surfaces and their interactions with aqueous solutions and microbial organisms. Chem. Rev. 99 77-174. [ Links ]
CARÉ S, NGUYEN QT, L'HOSTIS V and BERTHAUD Y (2008) Mechanical properties of the rust layer induced by impressed current method in reinforced mortar. Cement Concrete Res. 38 1079-1091. [ Links ]
CRAWFORD RJ, HARDING IH and MAINWARING DE (1993a) Adsorption and coprecipitation of single heavy metal ions onto the hydrated oxides of iron and chromium. Langmuir 9 3050-3056. [ Links ]
CRAWFORD RJ, HARDING IH and MAINWARING DE (1993b) Adsorption and coprecipitation of multiple heavy metal ions onto the hydrated oxides of iron and chromium. Langmuir 9 3057-3062. [ Links ]
CUNDY AB, HOPKINSON L AND WHITBY RLD (2008) Use of iron-based technologies in contaminated land and groundwater remediation: A review. Sci. Tot. Environ. 400 42-51. [ Links ]
DAVISON W and SEED G (1983) The kinetics of the oxidation of ferrous iron in synthetic and natural waters. Geochim. Cosmochim. Acta 47 67-79. [ Links ]
DIAO M and YAO M (2009) Use of zero-valent iron nanoparticles in inactivating microbes. Water Res. 43 5243-5251. [ Links ]
DOONG R, CHEN K and TSAI H-C (2003) Reductive dechlorination of carbon tetrachloride and tetrachloroethylene by zerovalent silicon-iron reductants. Environ. Sci. Technol. 37 2575-2581. [ Links ]
DUTRIZAC JE (1991) The precipitation of lead jarosite from chloride media. Hydrometal. 26 327-346. [ Links ]
ELSNER M, CWIERTNY DM, ROBERTS AL and LOLLAR BS (2007) Response to Comment on "1,1,2,2-Tetrachloroethane Reactions with OH-, Cr(II), Granular Iron, and a Copper-Iron Bimetal: Insights from Product Formation and Associated Carbon Isotope Fractionation". Environ. Sci. Technol. 41 7949-7950. [ Links ]
ERBS M, HANSEN HCB and OLSEN CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II) iron(III) hydroxide sulfate (green rust). Environ. Sci. Technol. 33 307-311. [ Links ]
GHAUCH A, ABOU ASSI H and TUQAN A (2010a) Investigating the mechanism of clofibric acid removal in Fe0/H2O systems. J. Hazard. Mater. 176 48-55. [ Links ]
GHAUCH A, ABOU ASSI H and BDEIR S (2010b) Aqueous removal of diclofenac by plated elemental iron: Bimetallic systems. J. Hazard. Mater. 182 64-74. [ Links ]
GILLHAM RW (2003) Discussion of nano-scale iron for dehalogenation. by Nyer E.K. and D.B. Vance. Ground Water Monit. Remed. 23 6. [ Links ]
GILLHAM RW and O'HANNESIN SF (1994) Enhanced degradation of halogenated aliphatics by zero-valent iron. Ground Water 32 958-967. [ Links ]
GU B, LIANG L, DICKEY MJ, YIN X and DAI S (1998) Reductive precipitation of uranium (VI) by zero-valent iron. Environ. Sci. Technol. 32 3366-3373. [ Links ]
HELLAND BR, ALVAREZ PJJ and SCHNOOR JL (1995) Reductive dechlorination of carbon tetrachloride with elemental iron. J. Hazard. Mater. 41 205-216. [ Links ]
HENDERSON AD and DEMOND AH (2007) Long-term performance of zero-valent iron permeable reactive barriers: a critical review. Environ. Eng. Sci. 24 401-423. [ Links ]
HUANG H and SORIAL GA (2007) Perchlorate remediation in aquatic systems by zero valent iron. Environ. Eng. Sci. 24 917-926. [ Links ]
HUSSAM A and MUNIR AKM (2007) A simple and effective arsenic filter based on composite iron matrix: Development and deployment studies for groundwater of Bangladesh. J. Environ. Sci. Health A 42 1869-1878. [ Links ]
JAMBOR JL, RAUDSEPP M and MOUNTJOY K (2005) Mineralogy of permeable reactive barriers for the attenuation of subsurface contaminants. Can. Miner. 43 2117-2140. [ Links ]
JEEN S-W, BLOWES DW and GILLHAM RW (2008) Performance evaluation of granular iron for removing hexavalent chromium under different geochemical conditions. J. Contam. Hydrol. 95 76-91. [ Links ]
JIAO Y, QIU C, HUANG L, WU K, MA H, CHEN S, MA L and WU L (2009) Reductive dechlorination of carbon tetrachloride by zerovalent iron and related iron corrosion. Appl. Catal. B: Environ. 91 434-440. [ Links ]
KAASALAINEN M and YLI-HALLA M (2003) Use of sequential extraction to assess metal partitioning in soils. Environ. Pollut. 126 225-233. [ Links ]
KANG S-H and CHOI W (2009) Response to comment on "Oxidative degradation of organic compounds using zero-valent iron in the presence of natural organic matter serving as an electron shuttle". Environ. Sci. Technol. 43 3966-3967. [ Links ]
KARTHIKEYAN KG, ELLIOTT HA and CANNON FS (1997) Adsorption and coprecipitation of copper with the hydrous oxides of iron and aluminum. Environ. Sci. Technol. 31 2721-2725. [ Links ]
KATSOYIANNIS IA, RUETTIMANN T and HUG SJ (2008) pH dependence of fenton reagent generation and As(III) oxidation and removal by corrosion of zero valent iron in aerated water. Environ. Sci. Technol. 42 7424-7430. [ Links ]
KEENEY-KENNICUTT WL and MORSE JW (1985) The redox chemistry of Pu(V)O2+ interaction with common mineral surfaces in dilute solutions and seawater. Geochim. Cosmochim. Acta 49 2577-2588. [ Links ]
KNOWLTON LG (1928) Some experiments on iron. J. Phys. Chem. 32 1572-1595. [ Links ]
KOMNITSAS K, BARTZAS G and PASPALIARIS I (2004) Efficiency of limestone and red mud barriers: laboratory column studies. Miner. Eng. 17 183-194. [ Links ]
KOMNITSAS K, BARTZAS G, FYTAS K and PASPALIARIS I (2007) Long-term efficiency and kinetic evaluation of ZVI barriers during clean up of copper containing solutions. Miner. Eng. 20 1200-1209. [ Links ]
KOMNITSAS K, BARTZAS G and PASPALIARIS I (2006) Modeling of reaction front progress in fly ash permeable reactive barriers. Environ. Forensics 7 219-231. [ Links ]
LAINE DF and CHENG IF (2007) The destruction of organic pollutants under mild reaction conditions: A review. Microchem. J. 85 183-193. [ Links ]
LANGMUIR D (1997) Aqueous Environmental Geochemistry. Prentice-Hall, Upper Saddle River. 600 pp. [ Links ]
LAVINE BK, AUSLANDER G and RITTER J (2001) Polarographic studies of zero valent iron as a reductant for remediation of nitroaromatics in the environment. Microchem. J. 70 69-83. [ Links ]
LEE G, RHO S and JAHNGD (2004) Design considerations for groundwater remediation using reduced metals. Korean J. Chem. Eng. 21 621-628. [ Links ]
LEE M, PAIK IS, KIM I, KANG H and LEE S (2007) Remediation of heavy metal contaminated groundwater originated from abandoned mine using lime and calcium carbonate. J. Hazard. Mater. 144 208-214. [ Links ]
LI T and FARRELL J (2000) Reductive dechlorination of trichloroethene and carbon tetrachloride using iron and palladized-iron cathodes. Environ. Sci. Technol. 34 173-179. [ Links ]
LI T and FARRELL J (2001) Electrochemical investigation of the ratelimiting mechanisms for trichloroethylene and carbon tetrachloride reduction at iron surfaces. Environ. Sci. Technol. 35 3560-3565. [ Links ]
LIEN H-L and WILKIN RT (2005) High-level arsenite removal from groundwater by zero-valent iron. Chemosphere 59 377-386. [ Links ]
LIPCZYNSKA-KOCHANY E, HARMS S, MILBURN R, SPRAH G and NADARAJAH N (1994) Degradation of carbon tetrachloride in the presence of iron and sulphur containing compounds. Chemosphere 29 1477-1489. [ Links ]
LIU X and MILLERO FJ (1999) The solubility of iron hydroxide in sodium chloride solutions. Geochim. Cosmochim. Acta 63 3487-3497. [ Links ]
MACKENZIE PD, HORNEY DP and SIVAVEC TM (1999) Mineral precipitation and porosity losses in granular iron columns. J. Hazard. Mater. 68 1-17. [ Links ]
MADRID F, REINOSO R, FLORIDO MC, DIAZ BARRIENTOS E, AJMONE-MARSAN F, DAVIDSON CM AND MADRID L (2007) Estimating the extractability of potentially toxic metals in urban soils: A comparison of several extracting solutions. Environ. Pollut. 147 713-722. [ Links ]
MANTHA R, TAYLOR KE, BISWAS N and BEWTRA JK (2001) A continuous system for Fe0 reduction of nitrobenzene in synthetic wastewater. Environ. Sci. Technol. 35 3231-3236. [ Links ]
MARTIN ST (2005) Precipitation and dissolution of iron and manganese oxides. In: Grassian VH (ed.) Environmental Catalysis. CRC Press, Boca Raton. 61-81. [ Links ]
MATHESON LJ and TRATNYEK PG (1994) Reductive dehalogenation of chlorinated methanes by iron metal. Environ. Sci. Technol. 28 2045-2053. [ Links ]
MENG Y-F, GUAN B-H, WU Z-B and WANG D-H (2006) Enhanced degradation of carbon tetrachloride by surfactant-modified zerovalent iron. J. Zhejiang Univ. Sci. B. 7 702-707. [ Links ]
MIELCZARSKI JA, ATENAS GM, MIELCZARSKI E (2005) Role of iron surface oxidation layers in decomposition of azo-dye water pollutants in weak acidic solutions. Appl. Catalysis B: Environ. 56 289-303. [ Links ]
MILLERO FJ, YAO W and AICHER J (1995) The speciation of Fe(II) and Fe(III) in natural waters. Mar. Chem. 50 21-39. [ Links ]
MORGAN B and LAHAV O (2007) The effect of pH on the kinetics of spontaneous Fe(II) oxidation by O2 in aqueous solution - basic principles and a simple heuristic description. Chemosphere 68 2080-2084. [ Links ]
NESIC S and LEE J K-L (2003) A mechanistic model for carbon dioxide corrosion of mild steel in the presence of protective iron. Carbonate films - Part 3: Film growth model. Corros. 59 616-628. [ Links ]
NESIC S, NORDSVEEN M, NYBORG R and STANGELAND A (2003) A mechanistic model for carbon dioxide corrosion of mild steel in the presence of protective iron carbonate films - Part 2: A numerical experiment. Corros. 59 489-497. [ Links ]
NESIC S (2007) Key issues related to modelling of internal corrosion of oil and gas pipelines - A review. Corros. Sci. 49 4308-4338. [ Links ]
NORDSVEEN M, NESIC S, NYBORG R and STANGELAND A (2003) A mechanistic model for carbon dioxide corrosion of mild steel in the presence of protective iron carbonate films - Part 1: Theory and verification. Corros. 59 443-456. [ Links ]
NOUBACTEP C, MEINRATH G, DIETRICH P and MERKEL B (2003) Mitigating uranium in ground water: prospects and limitations. Environ. Sci. Technol. 37 4304-4308. [ Links ]
NOUBACTEP C, SCHONER A and MEINRATH G (2006a) Mechanism of uranium (VI) fixation by elemental iron. J. Hazard. Mater. 132 202-212. [ Links ]
NOUBACTEP C, SCHONER A, DIENEMANN H and SAUTER M (2006b) Investigating the release of co-precipitated uranium from iron oxides. J. Radioanal. Nucl. Chem. 267 21-27. [ Links ]
NOUBACTEP C (2007) Processes of contaminant removal in "Fe0-H2O" systems revisited: The importance of co-precipitation. Open Environ. J. 1 9-13. [ Links ]
NOUBACTEP C (2008) A critical review on the mechanism of contaminant removal in Fe0-H2O systems. Environ. Technol. 29 909-920. [ Links ]
NOUBACTEP C and WOAFO P (2008) Elemental iron (Fe0) for better drinking water in rural areas of developing countries. In: Merkel BJ and Hasche-Berger A (eds.) Uranium in the Environment. Springer, Berlin, Heidelberg. 121-130. [ Links ]
NOUBACTEP C (2009a) On the operating mode of bimetallic systems for environmental remediation. J. Hazard. Mater. 164 394-395. [ Links ]
NOUBACTEP C (2009b) On the validity of specific rate constants (kSA) in Fe0/H2O systems. J. Hazard. Mater. 164 835-837. [ Links ]
NOUBACTEP C (2009c) Characterizing the discoloration of methylene blue in Fe0/H2O systems. J. Hazard. Mater. 166 79-87. [ Links ]
NOUBACTEP C (2009d) Metallic iron for environmental remediation: Learning from the Becher Process. J. Hazard. Mater. 168 1609-1612. [ Links ]
NOUBACTEP C (2009e) Characterizing the reactivity of metallic iron upon methylene blue discoloration in Fe0/MnO2/H2O systems. J. Hazard. Mater. 168 1613-1616. [ Links ]
NOUBACTEP C (2009f) An analysis of the evolution of reactive species in Fe0/H2O systems. J. Hazard. Mater. 168 1626-1631. [ Links ]
NOUBACTEP C (2009g) Characterizing the effects of shaking intensity on the kinetics of metallic iron dissolution in EDTA. J. Hazard. Mater. 170 1149-1155. [ Links ]
NOUBACTEP C and SCHONER A (2009) Fe0-based alloys for environmental remediation: Thinking outside the box. J. Hazard. Mater. 165 1210-1214. [ Links ]
NOUBACTEP C, KURTH A-MF and SAUTER M (2009a) Evaluation of the effects of shaking intensity on the process of methylene blue discoloration by metallic iron. J. Hazard. Mater. 169 1005-1011 [ Links ]
NOUBACTEP C, SCHONER A and WOAFO P (2009b) Metallic iron filters for universal access to safe drinking water. CLEAN - Soil Air Water 37 930-937. [ Links ]
NOUBACTEP C (2010a) The suitability of metallic iron for environmental remediation. Environ. Progr. Sust. En. 29 286-291. [ Links ]
NOUBACTEP C (2010b) Elemental metals for environmental remediation: Learning from cementation process. J. Hazard. Mater. 181 1170-1174. [ Links ]
NOUBACTEP C and CARE S (2010a) On nanoscale metallic iron for groundwater remediation. J. Hazard. Mater. 182 (1-3) 923-927. [ Links ]
NOUBACTEP C and CARE S (2010b) Enhancing sustainability of household water filters bymixing metallic iron with porous materials. Chem. Eng. J. 162 (2) 635-642. [ Links ]
NOUBACTEP C and CARE S (2010c) Dimensioning metallic iron beds for efficient contaminant removal. Chem. Eng. J. 163 454-460. [ Links ]
NOUBACTEP C and SCHONER A (2010a) Metallic iron for environmental remediation: Learning from Electrocoagulation. J. Hazard. Mater. 175 1075-1080. [ Links ]
NOUBACTEP C and SCHONER A (2010b) Metallic iron: dawn of a new era of drinking water treatment research? Fresen. Environ. Bull. 19 1661-1668. [ Links ]
NOUBACTEP C, SCHONER A and SAUTER M (2010) Significance of oxide-film in discussing the mechanism of contaminant removal by elemental iron materials. In: Kaneco S, Viswanathan B, Katsumata H (eds.) Photo-Electrochemistry and Photo-Biology for the Sustainablity. Bentham Science Publishers. 34-55. [ Links ]
O'HANNESIN SF and GILLHAM RW (1998) Long-term performance of an in situ "iron wall" for remediation of VOCs. Ground Water 36 164-170. [ Links ]
PALMER CD and WITTBRODT PR (1991) Processes affecting the remediation of chromium-contaminated sites. Environ. Health Perspect. 92 25-40. [ Links ]
POLASEK P (2007) Differentiation between different kinds of mixing in water purification. Water SA 33 249-252. [ Links ]
POST JE (1999) Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 96 3447-3454. [ Links ]
POSTMA D (1985) Concentration of Mn and separation from Fe in sediments - I. Kinetics and stoichiometry of the reaction between birnessite and dissolved Fe(II) at 10ºC. Geochim. Cosmochim. Acta 49 1023-1033. [ Links ]
POWELL MR, PULS WR, HIGHTOWER KS and SEBATINI AD (1995) coupled iron corrosion and chromate reduction: Mechanisms for subsurface remediation. Environ. Sci. Technol. 29 1913-1922. [ Links ]
RANGSIVEK R and JEKEL MR (2005) Removal of dissolved metals by zero-valent iron (ZVI): Kinetics, equilibria, processes and implications for stormwater runoff treatment. Water Res. 39 4153-4163. [ Links ]
REYNOLDS GW, HOFF JT and GILLHAM RW (1990) Sampling bias caused by materials used to monitor halocarbons in groundwater. Environ. Sci. Technol. 24 135 -142. [ Links ]
RIBA O, SCOTT TB, RAGNARSDOTTIR KV and ALLEN GC (2008) Reaction mechanism of uranyl in the presence of zero-valent iron nanoparticles. Geochim. Cosmochim. Acta 72 4047-4057. [ Links ]
RICKARD D (2006) The solubility of FeS. Geochim. Cosmochim. Acta 70 5779-5789. [ Links ]
SATOH Y, KIKUCHI K, KINOSHITA S and SASAKI H (2006) Potential capacity of coprecipitation of dissolved organic carbon (DOC) with iron(III) precipitates. Limnol. 7 231-235. [ Links ]
SCHERER MM, RICHTER S, VALENTINE RL and ALVAREZ PJJ (2000) Chemistry and microbiology of permeable reactive barriers for in situ groundwater clean up. Crit. Rev. Environ. Sci. Technol. 30 363-411. [ Links ]
SCHREIER CG and REINHARD M (1994) Transformation of chlorinated organic compounds by iron and manganese powders in buffered water and in landfill leachate. Chemosphere 29 1743-1753. [ Links ]
SCHWERTMANN U, WAGNER F and KNICKER H (2005) Ferrihydrite-humic associations magnetic hyperfine interactions. Soil Sci. Soc. Am. J. 69 1009-1015. [ Links ]
SCOTT NK, GREEN FJ, DO DH and MCLEAN JS (1995) Arsenic removed by coagulation. J. Am. Water Works Assoc. 87 114-126. [ Links ]
SHI Z and STONE AT (2009) PbO2(s, Plattnerite) Reductive dissolution by aqueous manganous and ferrous ions. Environ. Sci. Technol. 43 3596-3603. [ Links ]
SHI Z and STONE AT (2009) PbO2(s, Plattnerite) Reductive dissolution by natural organic matter: Reductant and inhibitory subfractions. Environ. Sci. Technol. 43 3604-3611 [ Links ]
SIBRELL PL, CHAMBERS MA, DEAGUERO AL, WILDEMAN TR and REISMAN DJ (2007) An innovative carbonate coprecipitation process for the removal of zinc and manganese from mining impacted waters. Environ. Eng. Sci. 24 881-896. [ Links ]
STIPP SLS, HANSEN M, KRISTENSEN R, HOCHELLA Jr. MF, BENNEDSEN L, DIDERIKSEN K, BALIC-ZUNIC T, LEONARD D and MATHIEU H-J (2002) Behaviour of Fe-oxides relevant to contaminant uptake in the environment. Chem. Geol. 190 321-337. [ Links ]
STONE AT (1987a) Microbial metabolites and the reductive dissolution of manganese oxides: Oxalate and pyruvate. Geochim. Cosmochim. Acta 51 919-925. [ Links ]
STONE AT (1987b) Reductive dissolution of manganese(III/IV) oxides by substituted phenols. Environ. Sci. Technol. 21 979-988. [ Links ]
STONE AT and MORGAN JJ (1984a) Reduction and dissolution of manganese(III) and manganese(IV) oxides by organics. 1. Reaction with hydroquinone. Environ. Sci. Technol. 18 450-456. [ Links ]
STONE AT and MORGAN JJ (1984b) Reduction and dissolution of manganese(III) and manganese(IV) oxides by organics: 2. Survey of the reactivity of organics. Environ. Sci. Technol. 18 617-624. [ Links ]
SUGIYAMA Y and KUMAGAI T (2001) Chemical characterization of organic carbon dissolved in natural waters using inorganic adsorbents. Anal. Sci. 17 77-82. [ Links ]
TESSIER A, CAMPBELL PGC and BISSON M (1979) Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem. 51 844-851. [ Links ]
THIRUVENKATACHARI R, VIGNESWARAN S and NAIDU R (2008) Permeable reactive barrier for groundwater remediation. J. Ind. Eng. Chem. 14 145-156. [ Links ]
TOKORO C, YATSUGI Y, SASAKI H and OWADA S (2008) A quantitative modeling of co-precipitation phenomena in wastewater containing dilute anions with ferrihydrite using a surface complexation model. Resourc. Proc. 55 3-8. [ Links ]
TRATNYEK PG and SALTER AJ (2010) Response to Comment on "Degradation of 1,2,3-Trichloropropane (TCP): Hydrolysis, Elimination, and Reduction by Iron and Zinc". Environ. Sci. Technol. 44 3198-3199. [ Links ]
VIOLANTE A, DEL GAUDIO S, PIGNA M, RICCIARDELLA M and BANERJEE D (2007) Coprecipitation of arsenate with metal oxides. 2. Nature, mineralogy, and reactivity of iron(III) precipitates. Environ. Sci. Technol. 41 8275-8280. [ Links ]
WANG J-l and CHENG R (2008) Reply to comments on "Comparison of reductive dechlorination of p-chlorophenol using Fe0 and nanosized Fe0", by C. Noubactep. J. Hazard. Mater. 150 850-851. [ Links ]
WEAVER RM and HOCHELLA (Jr.) MF (2003) The reactivity of seven Mn-oxides with Cr3+aq: A comparative analysis of a complex, environmentally important redox reaction. Am. Mineral. 88 2016-2027. [ Links ]
WEBER EJ (1996) Iron-mediated reductive transformations: investigation of reaction mechanism. Environ. Sci. Technol. 30 716-719. [ Links ]
WHITE AF and PETERSON ML (1996) Reduction of aqueous transition metal species on the surfaces of Fe(II)-containing oxides. Geochim. Cosmochim. Acta 60 3799-3814. [ Links ]
WILKIN RT and McNEIL MS (2003) Laboratory evaluation of zero-valent iron to treat water impacted by acid mine drainage. Chemosphere 53 715-725. [ Links ]
WILKIN RT, PULS RW and SEWELL GW (2003) Long-term performance of permeable reactive barriers using zero-valent iron: Geochemical and microbiological effects. Ground Water 41 493-503. [ Links ]
YOU Y, HAN J, CHIU PC and JIN Y (2005) Removal and inactivation of waterborne viruses using zerovalent iron. Environ. Sci. Technol. 39 9263-9269. [ Links ]
Received 1 December 2007; accepted in revised form 3 September 2010.
* To whom all correspondence should be addressed. +49 551 39 3191; fax: +49 551 399379; e-mail: cnoubac@gwdg.de

