










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.36 n.4 Pretoria Jul. 2010
Optimisation of the membrane-assisted passive sampler and its comparison with solid phase extraction technique
Hlengilizwe NyoniI; Luke ChimukaI,*; Branislav VranaII; Ewa CukrowskaI; Hlanganani TutuI
ISchool of Chemistry, University of the Witwatersrand, Private Bag 3, WITS 2050, Johannesburg, South Africa
IISlovak National Water Reference Laboratory, Water Research Institute, Nábr. arm. gen. L. Svobodu 5, 81249 Bratislava, Slovakia
ABSTRACT
A novel membrane-assisted passive sampler was further optimised in the laboratory. It was then compared to the solid phase extraction technique in terms of the extraction efficiency, enrichment factor, detection limit and selectivity in wastewater. The passive sampler was exposed to 3  wastewater samples under laboratory conditions for 3 days. Five hundred millilitres of wastewater was extracted with C18 cartridges. The extraction efficiency of the passive sampler ranged from 4 to 10% while in solid phase extraction it was 40 to 67% for the 3 chlorophenols. In both cases, extraction efficiency was highest for 2,4-dichlorophenol. The low extraction efficiency in the passive sampler supports the idea that it is not an exhaustive extraction technique and does not disturb the chemical equilibrium of the sample. It therefore measures the bioavailable fraction of the compound and can be used for equilibrium sampling and extraction. The obtained enrichment factors from the passive sampler were 89 and 295 for 2-chlorophenol and 2,4-dichlorophenol, respectively. From solid phase extraction, enrichment factors of 102, 113 and 167 were obtained for 2-chlorophenol, 4-chlorophenol and 2,4-dichlorophenol, respectively. The enrichment factor (~2.5) and sampling rates (~28 µ·h-1) were both low for 4-chlorophenol in wastewater from passive sampler extraction. The calculated sampling rates were found to be 2 604 µ·h-1 for 2-chlorophenol, 1 074 µ·h-1 for 4-chlorophenol and 5 089 µ·h-1 for 2,4-dichlorophenol in spiked deionised water. In wastewater, the sampling rates were found to be 1 544 µ·h-1 for 2-chlorophenol, 28 µ·h-1 for 4-chlorophenol and 5 106 µ·h-1 for 2,4-dichlorophenol. The passive sampler was found to be superior in its selectivity towards the target compounds compared to solid phase extraction technique with C18 sorbent. Chromatograms from solid phase extraction of wastewater contained high peaks of unidentified, potentially interfering compounds, especially in the early part of the chromatogram. In contrast, chromatograms from the passive sampler extraction were very clean. The detection limits of the passive sampler were comparable with that of solid phase extraction and were around 1.5 µg·-1 except for 4-chlorophenol that was high in wastewater (~100 µg·-1).
wastewater samples under laboratory conditions for 3 days. Five hundred millilitres of wastewater was extracted with C18 cartridges. The extraction efficiency of the passive sampler ranged from 4 to 10% while in solid phase extraction it was 40 to 67% for the 3 chlorophenols. In both cases, extraction efficiency was highest for 2,4-dichlorophenol. The low extraction efficiency in the passive sampler supports the idea that it is not an exhaustive extraction technique and does not disturb the chemical equilibrium of the sample. It therefore measures the bioavailable fraction of the compound and can be used for equilibrium sampling and extraction. The obtained enrichment factors from the passive sampler were 89 and 295 for 2-chlorophenol and 2,4-dichlorophenol, respectively. From solid phase extraction, enrichment factors of 102, 113 and 167 were obtained for 2-chlorophenol, 4-chlorophenol and 2,4-dichlorophenol, respectively. The enrichment factor (~2.5) and sampling rates (~28 µ·h-1) were both low for 4-chlorophenol in wastewater from passive sampler extraction. The calculated sampling rates were found to be 2 604 µ·h-1 for 2-chlorophenol, 1 074 µ·h-1 for 4-chlorophenol and 5 089 µ·h-1 for 2,4-dichlorophenol in spiked deionised water. In wastewater, the sampling rates were found to be 1 544 µ·h-1 for 2-chlorophenol, 28 µ·h-1 for 4-chlorophenol and 5 106 µ·h-1 for 2,4-dichlorophenol. The passive sampler was found to be superior in its selectivity towards the target compounds compared to solid phase extraction technique with C18 sorbent. Chromatograms from solid phase extraction of wastewater contained high peaks of unidentified, potentially interfering compounds, especially in the early part of the chromatogram. In contrast, chromatograms from the passive sampler extraction were very clean. The detection limits of the passive sampler were comparable with that of solid phase extraction and were around 1.5 µg·-1 except for 4-chlorophenol that was high in wastewater (~100 µg·-1).
Keywords: passive sampler, solid phase extraction, water monitoring, chlorophenols, selectivity
Introduction
The common approach in risk assessment of water bodies is to collect a sample of the water, which is then analysed for potential pollutants in the laboratory. This approach, among other advantages, provides manageable control over accuracy and precision of the results. However, information obtained from spot water samples is only about concentration levels at the time of sampling and may fail to account for episodic contamination. This can be addressed by collecting a series of samples over a time period to obtain a more representative picture of water quality, but the cost of analysis is then increased. An alternative and more cost-effective approach is to obtain time-weighted average (TWA) concentrations of pollutants using passive samplers. Passive samplers are now seen as an alternative to conventional spot samples. This is seen by a number of recent reviews on passive samplers (Vrana et al., 2005; Lu et al., 2002; Petty et al., 2000; Stuer-Lauridsen, 2005; Chimuka and Cukrowska, 2006; Kot-Wasik et al., 2007). One of the first samplers for the monitoring of compounds in water bodies was developed by Södergren (1987). It consists of a dialysis membrane filled with an organic solvent. The major disadvantage of the system was the successive loss of the organic solvent from the device through diffusion during environmental exposure, lack of selectivity as well as a low sensitivity due to a high mass transfer resistance in the cellulose membrane. Huckins et al. (1990) described the semi-permeable membrane device (SPMD) sampler for passive and integrative in situ monitoring of nonpolar water- and air-borne contaminants. The SPMD sampler consists of layflat polyethylene tubing containing a thin film of triolein, a high molecular weight neutral lipid. This is supplied by Environmental Science Technologies Inc. The other advantage, apart from being very rugged, is that high preconcentration factors of the analytes are obtained. These are determined by the partition coefficients into the lipid and exposure time (Sabaliunas and Sodergren, 1997).
The disadvantage of the SPMD is the laborious recovery of the analytes, usually consuming large volumes of organic solvents. This has led to development of other passive samplers based on solid phase material as trapping media. These include the Chemcatcher, which uses commercially available solid phase extraction C18 Empore disks as receiving phase (Kingston et al., 2000; Vrana et al., 2006a). The type of Empore disk can also be chosen according to the properties of the analytes of interest. A membrane enclosed sorptive coating (MESCO) passive sampler that uses a stir bar coated with poly(dimethylsiloxane) (PDMS) enclosed in a dialysis membrane bag as receiving phase has also been described (Vrana et al., 2001). The stir bar used as a receiving phase is identical to the one used in the stir bar sorptive extraction (SBSE) technique (Baltussen et al., 1999). It combines the advantages of the passive sampling approach with solventless preconcentration of organic solutes from aqueous matrices and subsequent thermal desorption of the sequestered analytes on line with capillary gas chromatography. It avoids the clean-up of extracts, required for other samplers, and the whole extract is injected into the analytical instrument. A new MESCO that uses a silicone collector, instead of a stirrer bar coated with PDMS, has been reported by Paschke et al. (2006). The silicone rods are later thermally desorbed into a gas chromatographic system, as in the first prototype of MESCO. A polar organic chemical integrative sampler (POCIS) has been described by Alvarez et al. (2004). It consists of a solid receiving phase material enclosed in microporous polyethersulfone diffusion membrane and is suitable for sampling of polar organic compounds such as polar industrial pollutants, pesticides, personal care products, pharmaceuticals, etc. Jonsson's research group (Liu et al., 2005) has also reported the development of an equilibrium sampling through membranes (ESTM) technique for measuring the free fraction of ionisable organic compounds in water that could be applied as a passive sampler. The same research group has recently reported an equilibrium sampling of freely dissolved organic chemicals into a thin film of 1-octanol supported on a porous hollow fibre membrane, which could be applied as a passive sampler (Liu et al., 2006). In both of the above cases, solvent loss may be a problem when the equilibrium extraction techniques are used as passive samplers.
In our previous work (Chimuka et al., 2008), we reported a simple and novel membrane assisted passive sampler based on thin-walled silicone tubing. The sampler does not use any organic solvents and is ideal for monitoring of ionisable organic compounds in water bodies. Some other important parameters that affect the sampler performance were optimised in the previous study (Chimuka et al., 2008). These are length and thickness of the hollow fibre, concentration of the acceptor buffer, sample volume and extraction time, influence of stirring and sample concentration. Detailed discussions about these are found in Chimuka et al. (2008). In brief, a minimum sample volume of 750 m was found optimal for a 3-day exposure period. A linear relationship was also found between a 3-day exposure time and an accumulated amount which is in agreement with other passive samplers such as the Chemcatcher (Kingston et al., 2000; Vran et al., 2006a) MESCO (Vrana et al., 2001; Vrana et al., 2006b) and PDMS (Paschke et al., 2006). This means that trapping conditions were correctly set in the acceptor phase (Chimuka et al., 1998; Jonsson and Mathiasson, 1990).
In this work, the membrane-assisted passive sampler has been optimised further, in particular for studying the influence of humic substances and temperature on the extraction process, using chlorophenols as model compounds. Chlorophenols were chosen because of their toxicity; persistence and their widespread release as byproducts in the production of plastics and dyes, pulp and paper industries. Chlorinated phenols can also form during wastewater treatment, since chlorine is added as a disinfectant. In this study, the membrane-assisted passive sampler (MAPS) was compared to the solid phase extraction technique in terms of extraction efficiency, enrichment factor, detection limit and selectivity in wastewater.
Materials and methods
Chemicals and solutions
4-chlorophenol (>99%), 2-chlorophenol (>98%) and 2,4-dichlorophenol (>99%) were provided by Merck-Schuchardt (Darmstadt, Germany). Other chemicals used were trisodium phosphate (99%), proanalysis sulphuric acid (99%) and HPLC grade methanol and acetonitrile, all from Merck-Schuchardt (Darmstadt, Germany).
Stock solutions of chlorophenol standards at 1 000 mg·-1 were prepared in methanol and stored in the refrigerator at 4ºC. Fresh stock solutions were prepared every 3 months. Standard solutions for calibration purposes were prepared by diluting the stock with deionised water. The basic acceptor solution for trapping chlorophenols was obtained by dissolving trisodium phosphate buffer in deionised water at concentrations of 0.5 M. The calibration curve used was also tested for linearity from 0.5 mg·-1 to 40 mg·-1. External calibration standards were prepared from the stock solutions in deionised water. The concentration of the phenols in the extracted samples was thus quantified by injecting along the calibration standard solutions. Peak areas were used for quantification. Standard solutions in the µg·-1 concentration range were prepared daily while those in the mg·-1 range were stored in the refrigerator for about 3 days.
Chromatographic conditions
The 3 chlorophenols were separated with a mobile phase composition of 60% water and 40% acetonitrile at a flow rate of 1.0 m·min-1 using HPLC with the UV detector set at 280 nm (Chimuka et al., 2008). A C18 column with dimensions 5 µm x 4 mm x 25 cm was used (Supelco, Bellefonte, PA, USA). An SRI (LA, California, USA) 210 HPLC system with a UV detector (VUV-24) and Peak Simple chromatographic data system was used. The mobile phase was degassed offline and filtered before use. The injection volume was 20 µ.
Hollow fibres
Hollow fibre silicone membranes used for the optimisation process were purchased from Technical Products Inc., (Georgia, USA). The hollow fibres were bought as long tubes and were cut to appropriate lengths (48 cm x 0.1575 cm I.D x 0.2413 cm O.D) giving a volume of the inner acceptor phase solution of approximately 1 000 µ when used. The length and thickness of the hollow fibre was previously optimised (Chimuka et al., 2008).
Preparation of the hollow fibre membranes and extraction procedure
The thin-walled silicone hollow-fibre membrane, previously soaked in deionised water, was filled with acceptor buffer using a 1 000 µ micropipette. The hollow fibre ends were tightened together and made in the form of a loop of about 3 cm in diameter. The outside was thoroughly rinsed with deionised water to remove any buffer spills. It was then hung inside an iron mesh (Fig. 1) and immersed in an appropriate 3 sample vessel for an appropriate time, usually 3 days. Sample exposure was performed without any stirring. Thereafter, it was taken out, the outside washed or flushed with deionised water and its contents transferred into a 4 m vial. The buffer solution of the acceptor solution was adjusted by adding 100 µ of 2 M sulphuric acid, giving a total volume of 1 245 µ. The extracts were either analysed immediately or stored in the refrigerator at 4ºC. Each experiment was repeated at least twice or thrice.
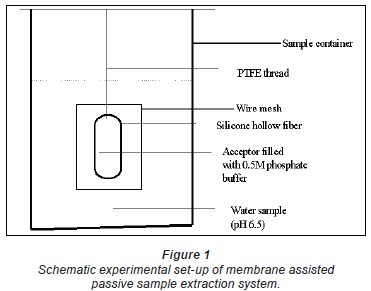
The extraction of chlorophenols in the sampler is similar to that for the supported liquid membrane (SLM) extraction technique (Chimuka et al., 1998; Jonsson and Mathiasson, 1999; Jonsson et al., 1993). In brief, the chlorophenols dissolve into the silicone membrane from the sample as non-ionised compounds. The compounds then diffuse through the membrane into the acceptor phase. Once in the acceptor phase, they are ionised and trapped. Because the pH of the acceptor phase solution is kept well above the pKa value of the chlorophenols, the concentration of non-ionised phenols in the acceptor phase is kept very close to zero. This maintains a concentration gradient between the 2 phases; the donor and acceptor phases. In this way the concentration of the compounds in the acceptor solution can be increased to much higher than in the original sample, without experiencing a plateau or maximum, and limited by the sample volume and/or the extraction time. This also gives selective enrichment since only compounds that are ionised at the pH of the acceptor phase are enriched (Megersa et al., 1999). Compounds that are ionised at the pH of the sample solution do not dissolve into the membrane since they are too polar. Neutral small compounds are not trapped in the acceptor phase while larger molecules tend to accumulate in the membrane and diffuse into the acceptor solution only slowly. Thus their sampling is limited due to very slow kinetics.
Optimisation experiments
Influence of the protective cover
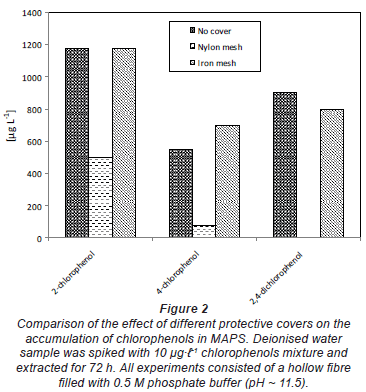
Experiments were conducted to study the influence of the protective cover of the sampler on the extraction process. For these experiments, the silicone hollow fibres were enclosed in a green nylon mesh and iron mesh. Each mesh was 9 cm (O.D) x 11 cm (length), with a pore size of 2 mm x 5 mm, and supported by an iron wire frame with about 1 mm thickness. A blank was also performed where no protective cover was used. Three litres of deionized water in stainless steel containers was used as a sample. The water was spiked with 10 µg·-1 of the mixture of chlorophenols. Exposure time was for 3 days, as described above, under static conditions.
Influence of temperature
To investigate the influence of temperature, sampler exposure was performed in the refrigerator at 4ºC, in a water bath at 16ºC and in a heated water bath at 40ºC. Before putting in the samplers, the spiked water samples were allowed to equilibrate for at least 2 h at the appropriate temperature. Deionised water containing 100 µg·-1 of each spiked chlorophenol was used as sample solution. The passive samplers were housed in an iron mesh. Passive sampler exposure period was 3 days. After extraction, the extracts were treated in the same way as described in the extraction procedure.
Studying the degree of trapping in the acceptor phase
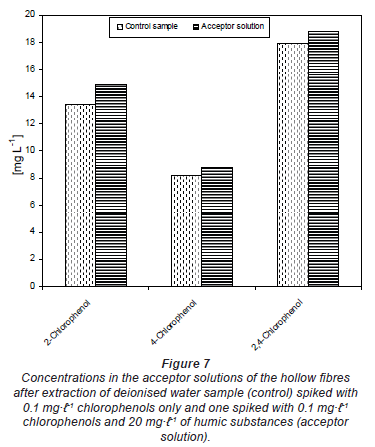
To study whether compounds trapped in the acceptor phase can diffuse back, a series of experiments was performed. The first series included spiking the acceptor buffer with about 1 mg·-1 of each chlorophenol. This was filled into the hollow fibre as before. The hollow fibres were then deployed in deionised and river water samples. The extraction was performed for 3 days. Following exposure, the contents of the acceptor solution were analysed to check for any loss of the chlorophenols from the acceptor solution. The same experiment was repeated but with deionised water spiked with 20 mg·-1 of humic substances. As a control, part of the spiked buffer solution was kept in the refrigerator at 4ºC during the entire period of extraction. This was also analysed at the same time as that of the acceptor solution from the hollow fibre.
Influence of humic substances in the sample
To study the effects of humic substances in the water on the performance of the passive sampler, deionised water containing 100 µg·-1 of each chlorophenol and 20 mg·-1 of humic substances was used as a sample solution. This sample solution was extracted with silicone hollow fibre membrane as before. Exposure period was 3 days. Afterwards, the enrichment factor was compared with those without any humic substances (in deionised water).
Reproducibility of the passive sampler
To test for reproducibility in wastewater and deionised water samples, 3 parallel passive samplers were deployed in each type of sample. The water samples were spiked with 100 µg·-1 of model compounds. From each experiment the enrichment factors were calculated together with the mean, standard deviation and per cent standard deviation.
Wastewater samples and solid phase extraction
To demonstrate the potential of the passive sampler, it was applied in the field at Goudkoppies wastewater treatment plant (GWWTP) west of Johannesburg. The GWWTP treats both household and industrial wastewater. Field parameters such as pH and conductivity were measured before deployment. The pH of the wastewater was found to be around 7.5. Passive samplers were deployed in the settling tanks and also after chlorination. Three parallel passive samplers were deployed at each site. The passive samplers in the settling tanks were tied to a beam rotating at 50 r/h. Wastewater samples were also collected by grab sampling at the 2 sites. The collected wastewater samples were also extracted using both the passive sampler under laboratory conditions and with the solid phase extraction technique. Some of the wastewater was spiked with 50 µg·-1 chlorophenols and extracted with both the passive sampler and the solid phase extraction technique. Wastewater samples that were extracted with solid phase extraction were filtered first and the pH adjusted to 4.5 with drops of 1 M nitric acid. Results of the determined concentrations of any chlorophenols in the blank and spiked samples were compared.
C18 solid phase extraction cartridges packed with 500 mg sorbent were used (Supelco, Park Bellefonte, USA). The extraction sequence was as follows: 6 m of methanol for conditioning followed by 6 m of deionised water for equilibration. Wastewater samples (500 m) adjusted to pH 4.5 with nitric acid were then passed at a flow rate of 5 m·min-1 through the solid phase extraction unit. The cartridge was washed with 3 m methanol-water (5:95 v/v). Elution was with 2 x 2 m methanol.
Calculations of extraction efficiency, enrichment factors and sampling rates
The extraction efficiency is defined as the fraction of analyte in the extracted sample that is found in the acceptor phase and is given by the equation below (Chimuka et al., 1998; Jonsson and Mathiasson 1999; Jonsson et al., 1993). It is also a measure of mass transfer between the donor and acceptor phase and is constant under specified extraction conditions.

where:
CA is the concentration in the collected acceptor fraction
CD is the concentration in the extracted sample
VA is the collected acceptor volume
VD is the volume of the sample that has been extracted.
The enrichment factor En is a ratio of concentration found in the acceptor phase to that in the original sample. This determines the detection limit of the method. It is given by the equation below (Chimuka et al., 1998; Jonsson and Mathiasson 1999; Jonsson et al., 1993; Megersa et al., 1999).

The amount of chemical accumulated in the sampler in the linear uptake phase is given by the following equation (Kingston et al., 2000; Vrana et al., 2001):
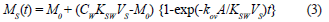
where:
MS is the mass of analyte in the receiving phase (acceptor phase)
M0 is the amount of analyte in the sampler at the start of exposure
CW is the water concentration of the analyte during deployment period and similar to CD
KSW is the equilibrium receiving phase/water distribution coefficient
A is the membrane surface area
kov is the overall mass-transfer coefficient
VS is the volume of the receiving phase (acceptor phase), similar to VA
t is the exposure time.
Results and discussion
Influence of the protective cover
Figure 2 gives the mean results of the optimisation of the protective cover of the sampler. The cover protects the sampler against small particles, including biofouling in water bodies. The results indicate that the type of cover is very important as it can affect the mass transfer process to the sampler. In this study, an iron mesh was found to be the best. Nylon mesh was found to reduce the mass transfer to the sampler, perhaps due to adsorption. As expected, the type of sample container does have an influence due to adsorption. Metal or glass containers are preferred when dealing with organic compounds.
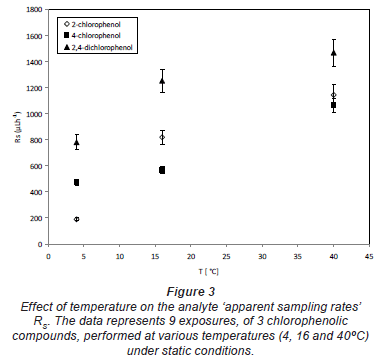
Influence of temperature
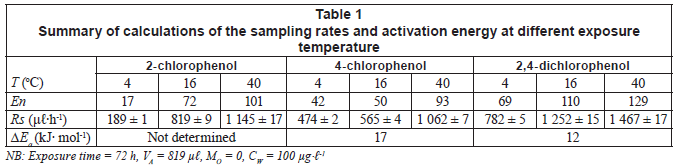
The relationship between sampling rates of 3 chlorophenolic compounds and temperature was compared at 3 temperatures (4, 16 and 40ºC). In general, the sampling rate increased with the increasing exposure temperature. The typical dependence of sampling rates on temperature is shown in Fig. 3.
Vrana et al. (2005) demonstrated that for the 4 polycyclic aromatic hydrocarbons with log KOW range from 4.0 to 5.1, the apparent receiving phase-water distribution coefficient was not significantly affected by temperature within the range from 6 to 18ºC. Consequently, the temperature was expected to affect mainly the magnitude of the kinetic component of the sampling rate.
Typically, it is quite clear that increased temperature of the environmental media can enhance mass transfer of compounds in all media. Thus for the 3 chlorophenols, namely 2-chlorophenol, 4-chlorophenol and 2, 4-dichlorophenol, with log KOW of 2.18, 2.39 and 2.96, respectively, the temperature dependence of the sampling rate RS can then be described by the Arrhenius-type equation:

where:
R is the universal gas constant (kJ·mol-1·K-1)
A is the pre-exponential factor expressing the maximum sampling rate at infinite temperature
T is the absolute temperature (K)
ΔEa is the activation energy (kJ·mol-1).
Values of ΔEa were obtained by plotting the natural logarithm of RS against the reciprocal value of absolute temperature (1/T) (Fig. 4). The intercept gives the value of ln A. The activation energy ΔEa can be calculated by multiplying the slope of the regression line (ΔEa/R) by R.
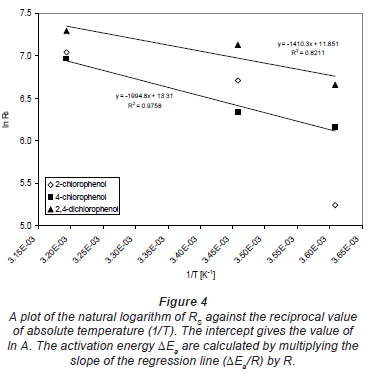
The sampling rates Rs obtained in the exposure experiments conducted at 100 µg·-1 water concentration and various temperatures are shown in Table 1.
The calculation of the activation energy ΔEa using Eq. (5) was performed on 3 sets of calibration data, obtained at stagnant water. The activation energies range between 12 and 17 kJ·mol-1. The average of all ΔEa values was 14 kJ·mol-1 with a standard deviation of 3.4 kJ·mol-1.
For a comparison, Vrana et al. (2005) calculated an average activation energy for Chemcatcher of 93 kJ·mol-1. The difference in activation energies is likely caused by the main resistance to mass transfer in the 2 samplers. This also depends on the polarity of the compounds. In a Chemcatcher, the main barrier to mass transfer for the PAHs studied is the aqueous boundary layer. In MAPS, the silicone layer also plays a critical role since analytes have to dissolve and diffuse through. Thus, the effect of temperature on the MAPS uptake kinetics appears to be less significant than that on Chemcatcher sampling rates. Note that the calculation of ΔEa was not performed for 2-chlorophenol because of very poor precision of the RS values.
The degree of trapping in the acceptor phase
The degree of trapping or ionisation of the target compounds in the acceptor phase is very important for quantification purposes of the sampler. This can also affect the exposure time. If all analytes in the acceptor phase are completely trapped, there is a linear relationship between extraction time and amount accumulated in the acceptor phase. A similar linear relationship also exists between the extracted concentration and the amount of the target compounds accumulated in the acceptor phase. From theory (Chimuka et al., 1998; Jonsson and Mathiasson 1999; Jonsson et al., 1993), once the pKa of the compound is known, it is possible to decide the pH of the acceptor solution at which all compounds will be almost or completely trapped. Co-extraction of other matrix compounds, especially if these are at high concentration, can lead to the acceptor solution reducing its pH value. This is due to neutralisation reactions of the acceptor solution with matrix components.
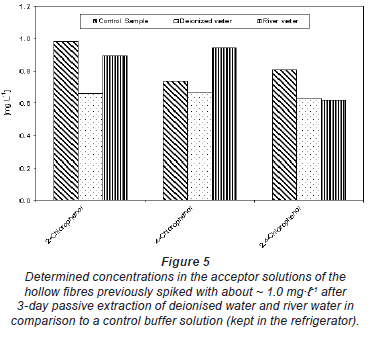
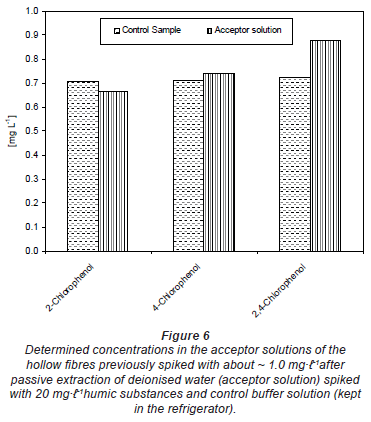
Figure 5 shows the concentration of chlorophenols determined in the acceptor solution of the hollow fibre after exposure to un-spiked deionised water (acceptor solution 1) and river water (acceptor solution 2). The acceptor solution was spiked with ~ 1.0 mg·-1 before filling it into the hollow fibre in each case. The control was the spiked buffer kept in the refrigerator during the passive sampler exposure period. The results in Fig. 5 generally indicate that there was no change in the spiked concentrations of the acceptor solution with time. The same results were obtained from deionised water spiked with 20 mg·-1 humic substances (Fig. 6). The differences seen in both Figs. 5 and 6 could be experimental error emanating from pH adjustment before HPLC analysis. This means that compounds were almost completely trapped in the acceptor solution and could not diffuse back and that the matrix component in river water or humic substances had no effect on the trapping capacity of the buffer solution.
Influence of humic substances in the sampler on the mass transfer
Figure 7 shows that there was no difference in the amount extracted in the acceptor solution of the passive samplers from a spiked deionised water sample and from one also containing humic substances. This means that the humic substances in the sampled water should not interfere with the extraction process of the chlorophenols under investigation. Humic substances may form weak or strong complexes with target compounds. This can reduce the dissolution of these compounds into the hollow fibre.
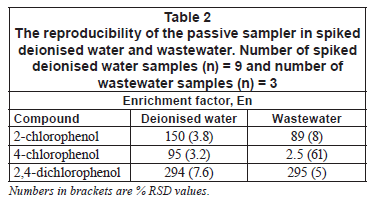
Reproducibility of the passive sampler
Table 2 indicates the reproducibility results of the passive sampler in deionised water and wastewater. The percentage relative standard deviations are typical of those found in other passive samplers under laboratory conditions (Vrana et al., 2005; Stuer-Lauridsen, 2005). For 4-chlorophenol, the percentage standard deviations were high in wastewater. This is due to the low extraction efficiency which was irreproducible. This suggests that the passive sampler behaves in the same manner as commercially available ones. The reproducibility under laboratory conditions also competes well with other active extraction techniques such as solid phase extraction.
Rate of accumulation
In applications of passive samplers, the sampling rate or rate of accumulation is used for quantification purposes. It is a measure of the mass transfer coefficient times sampler area (koxA) and is a constant at specified conditions. The rate of accumulation was also calculated directly from Eq. (3). Calculated apparent Rs values, from the results in Table 2, in deionised water are 2 604 µ·h-1 for 2-chlorophenol, 1 074 µ·h-1 for 4-chlorophenol and 5 089 µ·h-1 for 2,4-dichlorophenol. For wastewater, the apparent sampling rates from Table 2 are 1 544 µ·h-1 for 2-chlorophenol, 28 µ·h-1 for 4-chlorophenol and 5 106 µ·h-1 for 2,4-dichlorophenol.
The apparent rate of accumulation in wastewater for 4-chlorophenol is much lower than that found under comparable conditions in deionised water. This agrees well with the calculated enrichment factors. Vrana et al. (2006b) reported on a study of the sampling rates in a membrane-enclosed sorptive coating (MESCO) passive sampler for a variety of compounds. The sampling rates were found to differ by 10-fold and ranged from 100 to 983 µ·h-1, which is a narrow range in comparison to the broad Kow range of nearly 5 orders of magnitude for the compounds studied. The sampling rates obtained in our sampler in wastewater are slightly low compared to those in deionised water but are within the range of sampling rates found in the MESCO sampler. This means that the mass transfer in the passive sampler, especially through silicone rubber, into the acceptor phase is not slow.
Wastewater samples and comparison with solid phase extraction technique
There were no chlorophenols detected in any of the deployed passive samplers in the field after 3 days of deployment. Similarly, there were no chlorophenols detected from grab samples extracted by solid phase extraction and from the passive sampler under laboratory conditions. This means that concentration of the target compounds, if present in the wastewater, is at trace levels. Table 3 shows the obtained detection limits, which ranged from 1.00 to 100 µg·-1 for the passive sampler and from 1.80 to 2.45 µg·-1 for solid phase extraction. The detection limit was taken as the concentration that gives a signal to noise ratio of 3. The detection limits of the passive sampler after 3 days exposure is comparable to those obtained from solid phase extraction. Increasing the exposure time in the passive sampler from 3 days to 7 or more days can further lower the detection limit to sub-µg·-1 levels. This is equivalent to increasing the sample volume in solid phase extraction. For 4-chlorophenol, the detection limit was high. The same observation was obtained in the previous application of the passive sampler to river water (Chimuka et al., 2008). In deionised water, 4-chlorophenol (Table 2) was extracted well, but it was extracted poorly in wastewater, perhaps due to its interaction with matrix components.

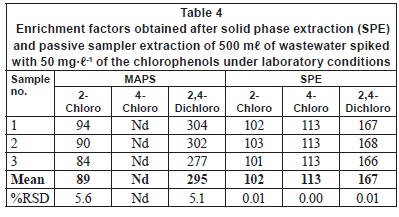
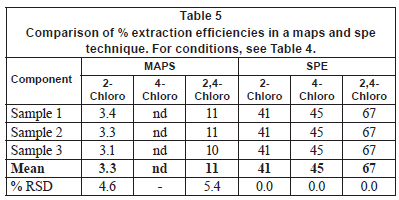
Tables 4 and 5 compare the obtained enrichment factors and extraction efficiencies of the chlorophenols using the passive sampler and solid phase extraction with spiked wastewater samples. The enrichment factors obtained with the passive sampler and solid phase extraction are comparable, as previously discussed for detection limits. Of more interest is the comparison of extraction efficiency between the passive sampler and solid phase extraction (Table 5). The extraction efficiency in the passive sampler was 3 and 11% for 2-chlorophenol and 2, 4-dichlorophenol, respectively. In solid phase extraction, the extraction efficiency was 41% and 67% for 2-chlorophenol and 2, 4-dichlorophenol, respectively. The low extraction efficiency in the passive sampler supports the idea that it is not an exhaustive extraction technique. The target analytes are not depleted in the bulk sample solution. The passive sampler therefore can be used as an equilibrium sampling and extraction technique. Equilibrium sampling and extraction does not change the chemical equilibrium of the components in the sample. This means that the passive sampler measures the truly dissolved bioavailable fraction of the chlorophenols. This is important in toxicity studies and ecological assessment of water bodies. Solid phase extraction, on the other hand, is an exhaustive technique and disturbs the chemical equilibrium of the sample. It therefore does not measure the truly bioavailable fraction of the compounds. The extraction efficiency in solid phase extraction was generally low, perhaps due to matrix components found in wastewater. It is common to have close to 100% extraction efficiency in solid phase extraction.
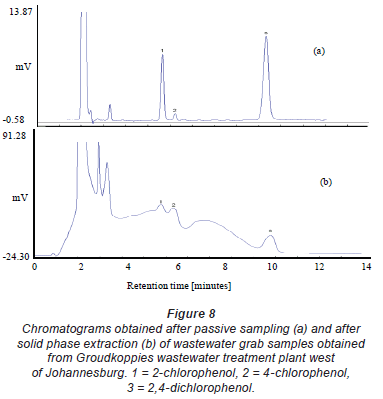
Another interesting comparison between the passive sampler and solid phase extraction is the selectivity. This is shown in the resulting chromatograms after passive sampling and solid phase extraction of wastewater (Fig. 8). From the passive sampler, a very clean chromatogram is obtained, whereas some matrix components are seen in a solid phase extraction chromatogram. The basis of the selectivity of the passive sampler has been discussed in detail in Chimuka et al. (2008). Most of the commercially available passive samplers are also not very selective since they use non-selective solid phase extraction sorbents as trapping media.
In solid phase extraction, each extraction consumed about 11 m of methanol in conditioning, rinsing and elution. For routine analysis, this adds up to sizeable amounts of organic solvents. In the developed passive sampler, no consumption of organic solvents is performed, which is an additional benefit of the sampler, besides its selectivity.
Conclusions
The type of protective cover does matter and can influence the mass transfer. Temperature was found to influence the mass transfer while humic substances did not. The membrane-assisted passive sample is comparable to solid phase extraction in terms of the most important parameters (enrichment factor, reproducibility and detection limits) that are needed for identification and quantification of target compounds in water under laboratory conditions. The passive sampler was found to be superior in selectivity towards the target compounds compared with solid phase extraction with C18 cartridges. The passive sampler also does not disturb the chemical equilibrium of the sample and therefore measures the bioavailable fraction during exposure as opposed to exhaustive solid phase extraction.
Acknowledgements
We acknowledge the financial support of the Slovak Research and Development Agency (Contract SK-ZA-0006-07), SA-National Research Foundation (Grant No. 67871) and Water Research Commission (Project No. K5/1504).
References
ALVAREZ DA, PETTY JD, HUCKINS JN, JONES-LEPP TL, GODDARD JP and MANAHAN SE (2004) Development of a passive, in situ, integrative sampler for hydrophilic organic contaminants in aquatic environments. Environ. Toxicol. Chem. 23 1640-1648. [ Links ]
BALTUSSEN E, SANDRA P, DAVID F and CRAMELS C (1999) Stir bar sorptive extraction (SBSE), a novel extraction technique for aqueous samples: Theory and principles. J. Microcolumn Sep. 11 737-747. [ Links ]
CHIMUKA L and CUKROWSKA E (2006) The role of passive samplers in monitoring aquatic ecosystems and occupational hygiene pollution. LC-GC North America 24 554-568. [ Links ]
CHIMUKA L, MEGERSA N, NORBERG J, MATHIASSON L and JONSSON, JA (1998) Incomplete trapping in supported liquid membrane extraction with a stagnant acceptor for weak bases. Anal. Chem. 70 3906-3911. [ Links ]
CHIMUKA L, NEMUTANDANI T, CUKROWSKA E and TUTU H (2008) Performance optimization of a membrane assisted passive sampler for monitoring of ionizable organic compounds in water. J. Environ. Monit. 10 129-135. [ Links ]
ENVIRONMENTAL SCIENCE TECHNOLOGIES INC. (USA) URL: http://est-lab.com (Accessed November 2009). [ Links ]
HUCKINS JN, TUBERGEN MW and MANUWEERA GK (1990) Semi permeable membrane devices containing model lipid: A new approach to monitoring the bioavailability of lipophilic contaminants and estimating their bioconcentration potential. Chemosphere 20 533-552. [ Links ]
JONSSON JA and MATHIASSON L (1999) Liquid membrane extraction in analytical sample preparation: II. Applications. Trends Anal. Chem. 18 325-334. [ Links ]
JONSSON JA, LOVKVIST P AUDUNSSON G and NILVÉ G (1993) Mass transfer kinetics for analytical enrichment and sample preparation using supported liquid membranes in a flow system with stagnant acceptor liquid. Analytica Chimica Acta 277 9-24. [ Links ]
KINGSTON JK, GREENWOOD R, MILLS GA, MORRISON GM and PERSSON LB (2000) Development of a novel passive sampling system for the time-averaged measurement of a range of organic pollutants in aquatic environments. J. Environ. Monit. 2 487-495. [ Links ]
KOT-WASIK A, ZABIEGALA B, URBANOWICZ M, DOMINIAK E, WASIK A and NAMIESNIK J (2007) Advances in passive sampling in environmental studies. Analytica Chimica Acta 602 141-163. [ Links ]
LIU JF, HU XL, PENG JF, JONSSON JA, MEYER P and JIANG GB (2006) Equilibrium sampling of freely dissolved alkyl phenols into a thin film of 1-octanol supported on a hollow fiber membrane. Anal. Chem. 78 8526-8534. [ Links ]
LIU JF, JONSSON JA and MAYER P (2005) Equilibrium sampling through membranes of freely dissolved chlorophenols in water samples with hollow fiber supported liquid membrane. Anal. Chem. 77 4800-4809. [ Links ]
LU Y, WANG Z and HUCKINS J (2002) Review of the background and application of triolein-containing semi permeable membrane devices in aquatic environmental study. Aquat. Toxicol. 60 139-153. [ Links ]
MEGERSA N, SOLOMON T and JONSSON JA (1999) Supported liquid membrane extraction for sample work-up and preconcentration of methoxy-s-triazine herbicides in a flow system. J. Chromatography A, 830 203-210. [ Links ]
PASCHKE A, SCHWAB K, BRUMMER J, SCHUURMANN G, PASCHKE H and POPP P (2006) Rapid semi-continuous calibration and field test of membrane enclosed silicone collector as passive water sampler. J. Chromatography A 1124 187-195. [ Links ]
PETTY JD, ORAZIO CE, HUCKINS JN, GALE RW, LEBO JA, MEADOWS JC, ECHOLS KR and CRANOR WI (2000) Considerations involved with the use of semi permeable membrane devices for monitoring environmental contaminants. J. Chromatography A 879 83-95 [ Links ]
SABALIUNAS P and SODERGREN A (1997) Use of semi-permeable membrane devices to monitor pollutants in water and assess their effects: A laboratory test and field verification. Environ. Pollut. 96 195-205. [ Links ]
SÖDERGREN A (1987) Solvent-filled dialysis membranes simulate uptake of pollutants by aquatic organisms. Environ. Sci. Technol. 21 855-859. [ Links ]
STUER-LAURIDSEN F (2005) Review of passive accumulation devices for monitoring organic micropollutants in the aquatic environment. Environ. Pollut. 136 503-524. [ Links ]
VRANA B, POPP P, PASCHKE A and SCHÜÜRMANN G (2001) Membrane-enclosed sorptive coating; An integrative passive sampler for monitoring organic contaminants in water. Anal. Chem. 73 5191-5200. [ Links ]
VRANA B, MILLS GA, ALLAN IJ, DOMINIAK E, SVENSSON K, KNUTSSON J, MORRISON G and GREENWOOD R (2005) Passive sampling techniques for monitoring pollutants in water. Trends Anal. Chem. 24 845-848. [ Links ]
VRANA B, MILLS GA, DOMINIAK E and GREENWOOD R (2006a) Calibration of the Chemcatcher passive sampler for the monitoring of priority organic pollutants in water. Environ. Poll. 142 333-343. [ Links ]
VRANA B, PASCHKE A and POPP P (2006b) Calibration and field performance of membrane-enclosed sorptive coating for integrative passive sampling of persistent organic pollutants in water. Environ. Pollut. 144 296-307. [ Links ]
Received $ February 2010; accepted in revised form 20 July 2010.
* To whom all correspondence should be addressed. +27(11) 717 6703, +27 (72) 497 8041; fax: (11) 717 6749; e-mail: luke.chimuka@wits.ac.za; luke_chimuka@hotmail.com


{kind=link}