










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.36 n.2 Pretoria Jan. 2010
Detection of Vibrio cholerae O1 in animal stools collected in rural areas of the Limpopo Province
V KeshavI; N PotgieterII; TG BarnardI, *
IWater and Health Research Unit, University of Johannesburg, PO Box 17011, Doornfontein 2028, South Africa
IIDepartment of Microbiology, Environmental Health, Domestic Hygiene and Microbial Pathogens Research Group, University of Venda, Private Bag X5050, Thohoyandou, 0950, Limpopo Province, South Africa
ABSTRACT
Vibrio cholerae (V. cholerae), the causative agent of cholera, has been responsible for various outbreaks worldwide and may be associated with animal faeces. In an attempt to understand the occurrence of this organism in the environment, 230 faecal samples were collected from pigs, chickens, goats, donkeys, cows and pigeons in rural areas of the Limpopo Province. Bacterial DNA was extracted from the faecal samples using a guanidium thiocyanate-based method. The DNA was screened for the presence of the sodB, rfb, FlaE, 16S rRNA and ctxA genes associated with V. cholerae, V. cholerae O1, V. cholerae O139 using 2 multiplex polymerase chain reactions (m-PCR). The V. cholerae sodB gene was detected in 74 of the 230 samples tested. Detection rates for the faecal samples obtained from individual species were as follows: cows (55/74), chickens (8/74), goats (2/74), donkeys (4/74), pigs (3/74) and pigeons (2/74). V. cholerae O1 was detected in (17/74) cow and (3/74) chicken samples, of which (9/17) cow samples and (3/3) chicken samples tested positive for toxigenic V. cholerae O1. The presence of this organism in faecal samples, taken close to water sources used by the villagers, raises the possibility that the causative V. cholerae O1 strain of the most recent outbreak in South Africa was present in the area 6 months prior to the outbreak.
Keywords: Vibrio cholerae, PCR, animal faeces, cholera toxin
Introduction
Cholera, caused by Vibrio cholerae, is a severe epidemic diarrhoeal disease which continues to devastate many developing countries where socioeconomic conditions are poor, sanitary systems and public hygiene are rudimentary, and safe drinking water is not available (Chen et al., 2007; Theron et al., 2000; Hunter, 1997). In rural areas of most developing countries people still use water from communal sources for domestic purposes. The sources may be unimproved (hand-dug wells, unprotected springs, rivers), with low and seasonal flow rates, or improved sources (public taps, borehole or pumps, protected wells, protected springs or harvested rainwater) (Gundry et al., 2006). Nevondo and Cloete (1999) reported that rivers in most rural areas of South Africa become polluted due to upstream activities such as washing clothes, bathing and animal activities and due to lack of sanitation.
V. cholerae has been isolated from surface water (Fraga et al., 2007; Percival et al., 2004) and the occurrence of V. cholerae in water sources can be linked to faecal pollution (Cox et al., 2005). Domestic and farm animals have been shown to be carriers of V. cholerae strains, contributing to their sustained presence in the area (Visser et al., 1999; Sanyal et al., 1974). Among the 193 currently recognised O serogroups of V. cholerae, only O1 and O139 have caused epidemics of cholera (Fraga et al., 2007). More than 95% of the strains belonging to serogroups O1 and O139 produce the cholera toxin (CT), which is central to the disease process (Chakraborty et al., 2000). Lipp et al. (2003) reported that 2 genes, rfb and wbe, are associated with the synthesis of the O-antigen in V. cholerae O1 and V. cholerae O139 and can be used to distinguish the 2 serogroups from each other. The genes coding for the cholera toxin can also be used to distinguish between toxigenic and non-toxigenic V. cholerae.
The polymerase chain reaction (PCR) is a rapid, oligonucleotide primer-directed in vitro method for replicating defined DNA sequences from target organisms (Brasher et al., 1998). PCR primers have been reported that allow for specific detection of a range of targets that include species-, serogroup- and virulence-specific genes. As a consequence of the speed, specificity and sensitivity of PCR, the procedure has become one of the most widely-used assays for direct detection of low levels of pathogenic microbes in environmental samples (Theron and Cloete, 2004). PCR is not dependent on the culturability of bacteria, and requires no additional conformational steps (Mumy and Findlay, 2004), leading to a significant decrease in sample analysis time.
The present study investigated the occurrence of Vibrio cholerae in animal faecal samples collected in rural areas in the Vhembe region of the Limpopo Province in South Africa using 2 multiplex PCRs.
Material and methods
Bacterial strains
The bacterial strains used during this study were obtained from the National Health Laboratory Services (NHLS) of South Africa, American Type Culture Collection (ATCC) and National Collection of Type Cultures (NCTC). All the bacterial strains were stored at -70°C in MicrobankTM cryovials (Pro-Lab Diagnostics). The strains were grown on nutrient agar (Oxoid®) at 37°C for 18 to 24 h.
Sample collection
A total of 230 faecal samples were randomly collected from the villages of the Vhembe region, during March and October 2008. The faecal samples were obtained from cows (147), chickens (38), donkeys (23), goats (10), pigs (6), dogs (4) and pigeons (2). The samples were collected aseptically as soon as possible after defecation and stored in sterile urine containers (Bioster) at 4°C until the samples were transported to the Water and Health Research Unit, Johannesburg Laboratory for analysis.
DNA extraction
An adaptation of the protocol reported by Boom et al. (1990) was used for this study. The changes included the use of the stool suspension as starting material. Approximately 0.15 g of each of the faecal samples was suspended in 1 mℓ phosphate buffered saline (pH 7.4; Sigma Aldrich, USA) containing 0.1% (vol/vol) Tween buffer (Merck, Germany) and homogenised by vortexing. Any debris that was not dissolved was removed by centrifugation for 15 s at 13 000 r/min, after which 200 uℓ of supernatant was transferred to a sterile 2 mℓ eppendorf tube to be used for the DNA extraction. To this, 700 µℓ L7A lysis buffer (120g guanidium thiocyanate, 0.1 M tris hydrochloride, 0.2 M EDTA, 2.6 g Triton X-100, 1 mg/ml ά-casein) was added and incubated at 70°C for 10 min. A volume of 250 µℓ 100% (vol/vol) ethanol was added to this mixture and further incubated at 56°C for 10 min. A celite solution (50 µℓ) was added and incubated at room temperature for 10 min (with occasional mixing). A sterile spin column prepared according to the method published by Borodina et al. (2003) was placed into a sterile 2 mℓ micro-fuge tube and the mixture loaded into the column. The mixture was loaded by adding approximately 500 µℓ into the column followed by centrifugation at 13 000 r/min for 30 s to separate the buffer from the celite. This step was repeated twice until all of the lysis mixture was loaded into the column. The column was washed twice with 400 µℓ wash buffer (120 g guanidium thiocyanate and 0.1 M tris hydrochloride ) and twice with 400 µℓ of a 70% (vol/vol (ethanol solution separating the liquid and solid phase each time by centrifugation at 13 000 r/min for 30 s. The last wash step was followed by a 2 min centrifugation step at 13 000 r/min to ensure that all the ethanol was removed from the column. The columns were transferred into clean sterile 1.5 mℓ micro-fuge tubes and 100 µℓ elution buffer (AE buffer, Qiagen) was added to the columns and incubated for 2 min at 56°C. The DNA was eluted from the columns by centrifugation for 2 min at 13 000 r/min, after which the columns were discarded. The DNA containing AE buffer was collected into the 1.5 mℓ microfuge tube. A negative control was included by performing the DNA extraction methods with only the DNA extraction reagents. A positive control was prepared by extracting DNA from 1.5 mℓ culture grown overnight suspended in sterile water.
PCR assays
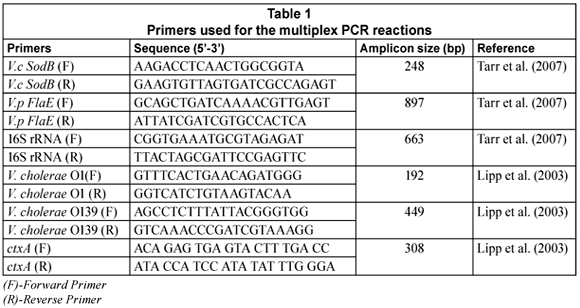
Two multiplex-PCR (m-PCR) assays were used for the detection and identification of V. cholerae species in the samples (Ntema, 2009). The 1st m-PCR targeted the sodB (V. cholerae species), FlaE (V. parahaemolyticus species) and 16S rRNA (Vibrio and Enterobacteriaceae species) genes (Tarr et al., 2007). The 2nd m-PCR targeted V. cholerae O1 and V. cholerae O139 rfb genes, ctxA (cholera toxin) and the 16S rRNA gene. For both the multiplex PCR's, the 16S rRNA primers were included as the positive internal control. The primers used for the amplification of these genes and the predicted sizes of the amplicons are shown in Table 1.
PCR reactions were performed in a Biorad MycyclerTM Thermal cycler in a total volume of 20 µℓ. Each reaction consisted of 10 µℓ 2x Qiagen m-PCR master mix (master mix contains HotStartTaq DNA Polymerase, m-PCR buffer with 2 mM MgCl2, and dNTP Mix); 1 µℓ 5x Qiagen Q-solution; 1-5 µℓ genomic DNA and PCR grade water. Two microlitres (2 µℓ) of the primer mix was used for the PCR reaction which consisted of 0.5 µℓ of a 10 mM stock of each V.c SodB primer; 1 µℓ of a 10 mM stock of each V.p FlaE primer and 0.16 µℓ of a 10 mM stock of each 16S rRNA primer (Table 1) for the 1st multiplex. The thermal cycling profile consisted of a 15 min enzyme activation step at 95°C followed by 35 cycles of denaturing at 92°C for 40 s, annealing at 57°C for 60 s, extension at 72°C for 90 s and a final elongation at 72°C for 7 min.
For the 2nd m-PCR, 0.25 µℓ of a 10 mM primer stock for each primer (V. cholerae O1, V. cholerae O139, ctxA, 16S rRNA) was added to the reaction mixture. Each reaction consisted of 10 µℓ 2x Qiagen m-PCR master mix 1 µℓ 5x Q-solution; 1-5 µℓ genomic DNA and PCR grade water. The m-PCR reactions were subjected to an enzyme activation step at 95°C for 15 min, followed by 35 cycles of denaturing at 94°C for 45 s, annealing at 55°C for 45 s, extension at 72°C for 60 s and a final elongation step at 72°C for 5 min.
Electrophoresis and visualisation of PCR products
DNA was analysed in a horizontal agarose slab gel (2.5% (weight/vol)) with ethidium bromide (0.5 µg/mℓ) in TAE buffer (40 mM tris acetate; 2 mM EDTA, pH8.3). The agarose gel was electrophoresed for 1 to 2 h at 80 to 100 V. The DNA was visualised with UV light (Gene Genius Bio Imaging System, Vacutec®). The relative sizes of the DNA fragments were estimated by comparing their electrophoretic mobility with 100 bp markers (Fermentas O' GeneRuler DNA ladder; Canada) that were run with the samples.
Results and discussion
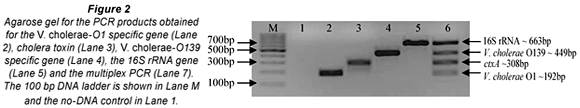
The aim of the study was to investigate the occurrence of V. cholerae species in animal faecal matter using PCR. To achieve this DNA was extracted from cow (147), chicken (38), donkey (23), goat (10), pig (6), dog (4) and pigeon (2) faecal samples and used as template for the multiplex PCR reactions. The m-PCR's were performed sequentially with all samples first tested for the presence of V. cholerae species specific sequences with the 1st m-PCR (Fig. 1). Samples that tested positive for the presence of V. cholerae species were subsequently tested for the presence of V. cholerae O1 or V. cholerae O139 specific O-antigen biosynthesis genes as well as the cholera toxin gene with the 2nd m-PCR (Fig. 2).

A total of 230 faecal samples were screened with the 1st m-PCR for the presence of V. cholerae (Table 2). Amplification of the 16S rRNA target from all samples indicated that bacterial DNA had been extracted and inhibitors had not suppressed amplification. The 16S rRNA primers included in the PCR targets the 16S rRNA gene of Enterobacteriaceae (Tarr et al., 2007) and were used as the internal control in the PCR.
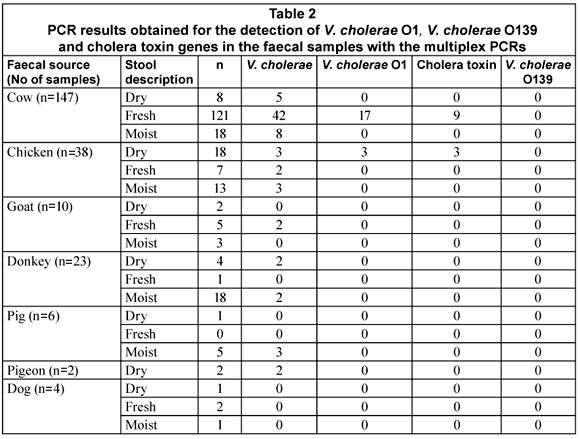
The V. cholerae sodB gene was detected in 32% (74/230) of the samples tested of which 74% (55/74) were cow faecal samples. The rest of the sodB positive samples were from chickens (8/74; 11%), goats (2/74; 3%), donkeys (4/74; 5%), pigs (3/74; 4%) and pigeons (2/74; 3%). Although the sodB gene was not detected in all of the samples, it was present in samples from all of the types of animals targeted, with the single exception of dogs. The V. cholerae O1 rfb gene was detected in only 27% (20/74) of the sodB positive samples and was restricted to samples obtained from cows (17/20; 85%) and chickens (3/20; 15%). Only (9/17; 53%) of the V. cholerae O1 positive cow samples showed the presence of the gene encoding the cholera toxin whereas all of the V. cholerae O1 positive chicken samples tested positive for the cholera toxin. No Vibrio cholerae O139 was detected in any of the samples.
Contrary to what would be expected, the occurrence of the V. cholerae in the samples could not be linked to the freshness of the faecal sample. V. cholerae was detected more often in fresh cow faecal samples compared to chickens where the V. cholerae was detected more in the dry faecal samples, especially V. cholerae O1. No conclusion about the survival and occurrence of V. cholerae can, however, be drawn at this stage, as factors such as sample collection during different seasonal conditions, animal age and herd immunity can all have an effect on the occurrence and detection of the organism (Cox et al., 2005).
The detection of V. cholerae in the animal faeces collected confirms reports that V. cholerae do occur in cow, chicken, dog and pig faecal samples (Visser et al., 1999; Sanyal et al., 1974). The one drawback experienced with this study was that although V. cholerae was detected nothing can be said about the viability of the organisms. PCR amplifies fragments of DNA that are present in dead and viable cells making it impossible to comment on the viability of the organism detected (Mieta, 2009).
Conclusion
V. cholerae, as well as V. cholerae O1, was detected in faecal samples obtained from animals in rural areas in the Vhembe region of the Limpopo Province, using a PCR-based technique. In most cases the animals were free to roam the area, including the areas around rivers and springs that are used as drinking water sources for some of the villages. Since V. cholerae could be detected in the animal faeces it can be concluded that the organism could find its way into the water sources as reported by Cox et al. (2005). Interestingly this area was part of the 2008/2009 cholera outbreak that started in Zimbabwe and spread to this province. This raises the possibility that the causative agent for the outbreak was introduced into the area from Zimbabwe or that it could have been present in the area 6 months before the outbreak started, during the time that the samples were collected.
It is important that attention is paid to the occurrence of V. cholerae O1 in animal faeces and to its possible health impact in an area where animal faeces are collected and used for agriculture, as well as construction and decorative purposes, in homes. Mpazi and Mntika (2005) reported that the spread and persistence of V. cholerae in rural areas can be attributed to the belief held by villagers that V. cholerae cannot be transmitted through cow and chicken faeces. It is therefore important that these uses of animal faeces should be kept in mind and included in analysis during diarrhoeal outbreaks.
References
BOOM R, SOL CJA, SALIMANS MMM, JANSEN CL, WERTHEIM-VAN DILLEN PME and VAN DER NOORDA J (1990) Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 28 (3) 495-503. [ Links ]
BORODINA TA, LEHRACH H and SOLDATOV AV (2003) DNA purification on homemade silica spin-columns. Anal. Biochem. 321 135-137. [ Links ]
BRASHER CW, DEPAOLA A, JONES DD and BEJ AK (1998) Detection of microbial pathogens in shellfish with mutliplex PCR. Curr. Microbiol. 37 101-107. [ Links ]
CHAKRABORTY S, MUKHOPADHYAY AK, BHADRA RK, GHOSH AN, MITRA R, SHIMADA T, YAMASAK S, FARUQUE SH, TAKED Y, COLWELL RR and NAIR GB (2000) Virulence genes in environmental strains of Vibrio cholerae. Appl. Environ. Microbiol. 66 (9) 4022-4028. [ Links ]
CHEN Y, JOHNSON JA, PUSCH GD, MORRIS JG and STINE OC (2007) The genome of non-O1 Vibrio cholerae NRT36S demonstrate the presence of pathogenic mechanisms that are distinct from those of O1 Vibrio cholerae. Infect. Immun. 75 (5) 2645-2647. [ Links ]
COX P, GRIFFITH M, ANGLES M, DEERE D and FERGUSON C (2005) Concentrations of pathogens and indicators in animal faeces in the Sydney Watershed. Appl. Environ. Microbiol. 10 5929-5934. [ Links ]
FRAGA SG, PICHEL M, COSTAGLIOLA M, CECILIA M, JURQUIZA V, PERESUTI S, CAFFER MI, AULET O, HOZBOR C, TRACANNA BC, DE GAMUNDI AV, HERNANDEZ D and RAMIREZ FC (2007) Environment and virulence factors of Vibrio cholerae strains isolated in Argentina. J. Appl. Microbiol. 103 (6) 2448-2456. [ Links ]
GUNDRY SW, WRIGHT JA, CONROY R, DU PREEZ M, GENTHE B, MOYO S, MUTISI C, NDAMBA J and POTGIETER N (2006) Contamination of drinking water between source and point-of-use in rural households of South Africa and Zimbabwe: Implications for monitoring the millennium development goal for water. Water Pract. Technol. 1 1-9. [ Links ]
HUNTER PR (1997) Waterborne Disease: Epidemiology and Ecology. John Wiley and Sons. UK. [ Links ]
LIPP EK, RIVERA ING, GIL AI, ESPELAND EM, CHOOPUN N, LOUIS VR, RUSSEK-COHEN E, HUQ A and COLWELL RR (2003) Direct detection of Vibrio cholerae and ctxA in Peruvian coastal water and plankton by PCR. Appl. Environ. Microbiol. 69 3676-3680. [ Links ]
MIETA S (2009) Detection of Selected Entero-Pathogenic Bacteria from Stool Specimens Using a Novel Collection Technique. Master of Technology Dissertation, University of Johannesburg, South Africa. [ Links ]
MPAZI VM and MNYIKA KS (2005) Knowledge, attitudes and practices regarding cholera outbreaks in Ilala Municipality of Dar Es Salaam Region, Tanzania. East Afr. J. Public Health 2 6-11. [ Links ]
MUMY KL and FINDLAY RH (2004) Convenient determination of DNA extraction efficiency using an external DNA recovery standard and quantitative-competitive PCR. J. Microbiol. Methods 57 259-268. [ Links ]
NEVONDO TS and CLOETE TE (1999) Bacterial and chemical quality of water supply in the Dertig village settlement. Water SA 25 215-220. [ Links ]
NTEMA VM (2009) Detection of Vibrio cholerae and Vibrio parahaemolyticus by Molecular and Culture Methods from Source Water to Household Container-Stored Water at the Point-Of-Use in Rural Vhembe Communities in South Africa. Master of Technology Dissertation, University of Johannesburg, South Africa. [ Links ]
PERCIVAL SL, CHALMERS RM, EMBREY M, HUNTER PR, SELLWOOD J and WYN-JONES P (2004) Microbiology of Waterborne Diseases. Elsevier Academic Press. UK. [ Links ]
SANYAL SC, SINGH SJ, TIWARI IC, SEN PC, MARWAH SM, HAZARIKA UR, SINGH H, SHIMADA T and SAKAZAKI R (1974) Role of Household Animals in Maintenance of Cholera Infection in a Community. J. Infect. Dis. 130 (6) 575-579. [ Links ]
TARR CL, PATEL JS, PUHR ND, SOWERS EG, BOPP CA and STROCKBINE NA (2007) Identification of Vibrio isolates by a multiplex PCR assay and rpoB sequence determination. J. Clin. Microbiol. 45 (1) 145-140. [ Links ]
THERON J, CILLIERS J, DU PREEZ M, BROZEL VS and VENTER SN (2000) Detection of toxigenic Vibrio cholerae from environmental water samples by an enrichment broth cultivation-pit-stop semi-nested PCR procedure. J. Appl. Microbiol. 89 539-546. [ Links ]
THERON J and CLOETE TE (2004) Emerging microbiological detection techniques. In: In: Cloete TE, Rose J, Nel LH and Ford T (ed.). Microbial Waterborne Pathogens. IWA Publishing, UK. [ Links ]
VISSER IJ, VELLEMA P, VAN DOKKUM H and SHIMADA T (1999) Isolation of Vibrio cholerae from diseased farm animals and surface water in The Netherlands. Vet. Rec. 144 (16) 451-452. [ Links ]
This paper was originally presented at the 2010 Southern African Young Water Professionals Conference, Pretoria, 19-20 January 2010
* To whom all correspondence should be addressed
+2711 559 6567; fax: +2711 559 6329
e-mail: tgbarnard@uj.ac.za

