










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.36 n.1 Pretoria Jan. 2010
Cr(VI) generation during sample preparation of solid samples – A chromite ore case study
RI GlastonburyI; W van der MerweI; JP BeukesI,*; PG van ZylI; G LachmannI; CJH SteenkampI; NF DawsonII; HM StewartII
IChemical Resource Beneficiation, North-West University, Potchefstroom Campus, Private Bag X6001, Potchefstroom 2520, South Africa
IIXstrata Alloys, PO Box 2131, Rustenburg 0300, South Africa
ABSTRACT
South Africa holds more than 70% of the world's viable chromite ore reserves and produces ~46.2% of the world's high carbon ferrochrome. It was recently reported that beneficiated South African chromite ores contained significant amounts of Cr(VI). If this is true, it could have serious consequences for South African chromite mines and the local environment. Currently none of these mines make any provision for Cr(VI) leaching from their mined ores. The data obtained in this study proved that the Cr(VI) content of chromite samples is influenced by the sample preparation technique employed prior to chemical analysis, more specifically, that pulverising of chromite samples in a normal atmospheric environment resulted in Cr(VI) formation. No Cr(VI) was liberated when pulverising was conducted in an inert atmosphere. The presence of Cr(VI) in South African chromite ores therefore seems unlikely. The results also suggest that the perceived threat of Cr(VI) contamination of groundwater and surface water, originating from chromite ore stockpiles, is improbable.
Keywords: hexavalent chromium, Cr(VI), sample preparation, pulverising, chromite.
Introduction
Chromium generally exists in 2 oxidation states in the environment, i.e. Cr(III) and Cr(VI). Cr(VI) is generally regarded as a carcinogenic, whereas Cr(III) is not (IARC, 1997). It is therefore important to differentiate between these 2 common oxidation states of chromium.
Since its discovery in 1798, chromite has remained the only commercially-recoverable source of chromium (Niagru, 1998; Cowey, 1994; Riekkoal-Vanhanen, 1999). It is generally accepted that South Africa holds between 74% (Cowey, 1994) and 80% (Riekkoal-Vanhanen, 1999) of the world's viable chromite ore reserves. The South African chromite reserves are situated within the Bushveld Complex (Howat, 1994). This geological phenomenon consists of an enormous saucer-like intrusive igneous mass, which extends for about 400 km from east to west and roughly the same distance from north to south. It is located in the central and western parts of the South African Highveld (Howat, 1994).
Chromite is primarily utilised for the production of ferrochrome, which is a crude alloy produced during the pyrometallurgical carbo-thermic reduction of chromite (Riekkoal-Vanhanen, 1999). Ferrochrome is mostly utilised for the manufacturing of stainless steel, which is a very important alloy in modern-day living. According to the 2007 production statistics of the International Chromium Development Association, South Africa produced approximately 46.2% of the world's Charge Chrome (which is the most common ferrochrome grade, containing typically 48.5 to 53.5% chromium) (ICDA, 2008).
It is commonly assumed that chromite contains only Cr(III) (Gu and Wills, 1988). In a recent article by Mandiwana et al. (2007), the Cr(VI) contents of samples collected from different stages of the production process at a ferrochrome smelter in South Africa were reported. It was found that 'chromium ore' contained 0.38 to 0.44 µg·g-1 of Cr(VI), while 'lumpy ore' contained 0.62 to 0·76 µg.g-1 (Mandiwana et al., 2007). Although the actual meaning of the term 'chromium ore' was not specified by the above-mentioned authors, it can be assumed that metallurgical grade ore was implied. Due to the friability of the South African chromite ores, it is common to only recover 10 to 15% lumpy ore (15 mm < typical size range < 150 mm) and 8 to 12% chip/pebble ores (6 mm < typical size range < 15 mm) during the beneficiation process employed after chromite mining. The remaining ore would typically be in the < 6 mm fraction, which would usually be crushed and/or milled to < 1 mm and then upgraded utilising typical gravity separation techniques (e.g. spiral concentrators) to ~45% Cr2O3 content. This upgraded < 1 mm ore is commonly known as metallurgical grade chromite ore.
If hexavalent chromium is present in South African chromite ore, it could have serious consequences for chromite mines and the local environment. None of these mines currently make any provision for water-soluble Cr(VI) leaching from their ores. The beneficiation processes employed (e.g. spiral concentration) are water-intensive and would contribute to the mobilisation of soluble hazardous compounds, if present. After beneficiation, the ore stockpiles are not placed on any specially constructed lining systems that would prevent leaching of hazardous water-soluble materials such as Cr(VI) into the groundwater.
However, in the above-mentioned paper (Mandiwana et al., 2007) it is stated that 'The samples were air-dried and homogizised by grinding in a IKA A11 milling system to a grain size less than 200 µm.' It has previously been reported (Beukes and Guest, 2001) that Cr(VI) can be generated during dry milling of chromium-containing materials, such as chromite. This has led to the suspicion that the sample preparation method used by Mandiwana et al. (2007) could have resulted in a bias towards recording Cr(VI) levels that were too high. Taking the size and importance of the South African chromite industry, as well as the pollution potential into consideration, the possible Cr(VI) content of local chromite ores is a significant issue and certainly needs verification. This prompted us to investigate:
• Whether the Cr(VI) contents reported in the South African chromite ores (Mandiwana et al., 2007) were generated during sample preparation, or whether Cr(VI) was already present in the chromite prior to sample preparation.
• Pulverising as a sample preparation technique, with specific reference to the likelihood of Cr(VI) generation. Pulverising is currently regarded as a standard step in sample preparation of solid samples, prior to chemical analysis by methods requiring solubilisation of the analyte.
Experimental
Materials
General
All chemicals used were analytical grade (AR) reagents, obtained from the different suppliers and used without any further purification. Standard Cr(VI) solutions were prepared from a 1 000 mg·ℓ-1 aqueous chromate analytical solution (Spectrascan, distributed by Teknolab AB, Sweden) and used for the calibration and verification of the analytical techniques employed. Phosphoric acid (B&M Scientific) and s-diphenyl carbazide (FLUKA) were used during Cr(VI) determination. Solutions of sodium hydroxide (Merck) and perchloric acid (Merck) were used to adjust the pH of aqueous solutions. Ultra-pure water (resistivity 18.2 MΩ·cm-1), produced by a Milli-Q water purification system, was used for all procedures requiring water. Liquid nitrogen was supplied by Afrox.
Chromite ore
Metallurgical-grade chromite ore samples (< 1 mm) were collected during a sampling campaign at the Xstrata Thorncliffe Mine in the Mpumalanga Province of South Africa. This mine is situated on the eastern limb of the Bushveld Complex, near the towns of Lydenburg, Steelpoort and Burgersfort (Howat, 1994). Thorncliffe typically supplies chromite to several ferrochrome smelters in the area. These samples were collected from stockpiles designated for ferrochrome smelters and can therefore be considered typical of ores utilised in the South African ferrochrome industry.
Methods
Pulverising experiments
A Siebtechnik pulveriser, commonly used to pulverise solid samples prior to chemical analysis, was used to conduct all pulverising experiments. All parts of the pulveriser which made contact with the actual chromite ore were made of tungsten carbide. This prevented possible iron contamination of the pulverised samples. It is well-known that metallic iron particles can reduce Cr(VI) to Cr(III). The metallurgical-grade ore used during pulvizising experiments was dried at 40ºC for 1 d and then cooled in airtight containers to avoid possible water absorption. The ore was stored in these containers and only removed prior to pulverising experiments. In all experiments 170 g of dry metallurgical grade chromite ore was pulverised. After a specific grind was completed, the contents of the pulveriser were collected in a sample bag for analysis. The pulveriser was then cleaned before the next grind commenced.
Particle size analysis
A Malvern Mastersizer 2000 was used to determine the particle size distribution of the pulverised chromite. A suspension of milled ore was ultra-sonicated for 60 s prior to the particle size measurement, in order to disperse the individual particles and to avoid the use of a chemical dispersant.
Cr(VI) extraction
The objective of Cr(VI) extraction should be to quantitatively extract Cr(VI), without inducing changes in speciation, i.e. Cr(III)/Cr(VI) inter-conversions (Pettine and Capri, 2005). The reviews by Ashley et al. (2003), Broadhurst and Maizda (2006) and Pettine and Capri (2005) provide recommendations with regard to the extraction of Cr(VI) from solid samples. For the determination of water-soluble and partially water-soluble Cr(VI) compounds, a buffered hotplate extraction with (NH4)2SO4–NH4OH seems to be the preferred option. A buffered hotplate extraction with Na2CO3–NaOH, performed in the presence of Mg(OH)2 or a purged N2 environment is recommended for extraction of total Cr(VI). The added Mg(OH)2, or purged N2 environment, is required to prevent Cr(III) oxidation. The Na2CO3–NaOH extraction would therefore have been ideal for this study, since it would have quantitatively extracted all Cr(VI) without inducing changes in the speciation. However, it was observed that this highly basic extraction medium interfered with the analytical technique employed (see next section), as small air bubbles tend to form inside the quartz cuvettes during uv/visible analysis. This can probably be attributed to the very acidic nature of the s-diphenyl carbazide (DPC) method (Pettine and Capri, 2005; Ashley et al., 2003), as opposed to the proposed ultra-basic extraction (Na2CO3–NaOH).
In order to overcome the above-mentioned problem, 2 very simple extraction methods, not involving any of the above-mentioned buffers, were employed. These were:
• 5 g of the solids were stirred for 2 h at room temperature in 70 mℓ water in a 100 mℓ glass beaker covered with parafilm to prevent material loss. Thereafter the leach water was filtered off by milli-pore filtering (0.45 µm) and the remaining solid residue was washed with 30 mℓ water. Leach water and wash water were combined in a 100 mℓ volumetric flask, which was filled to the calibration mark. This is the simplest Cr(VI) extraction method and is similar to the method applied by Beukes and Guest (2001).
• 5 g of the solids were stirred for 2 h at room temperature in 70 mℓ de-oxygenated pH 9 water. The water was de-oxygenated by bubbling gaseous N2 through it for 30 min prior to the extraction commencing. Extraction was conducted in a 100 mℓ round-bottom flask, while N2 bubbling was continued. Nitrogen de-oxygenation prevented Cr(III) oxidation during extraction. A higher than neutral pH was chosen in order to extract more Cr(VI), thus giving a closer resemblance to the basic extraction method as recommended by Ashley et al. (2003), as well as Broadhurst and Maidza (2006). However, the basicity of the extracting solution was not so high as to interfere with the analytical technique, as indicated previously. After the extraction was completed the leachate was filtered off by milli-pore filtering (0.45 µm) and the remaining solid residue was washed with 30 ml pH 9 de-oxygenated water. The wash water was combined with the leach water in a 100 mℓ volumetric flask, which was filled to the calibration mark with pH 9 de-oxygenated water.
Since neither of the above-mentioned extraction methods was conducted in ultra-basic aqueous environments, it could be expected that not all Cr(VI) containing salts would quantitatively be extracted. Therefore, Cr(VI) values reported in this paper should not be considered to represent the total Cr(VI) quantitatively. However, absolute total Cr(VI) concentrations were not considered as a pre-requisite for this work, since the main aim was to determine Cr(VI) generation trends.
Cr(VI) determination
The Cr(VI) content of the extracted solutions was determined with a Pharmacia Biotech Ultrospec 3 000 uv/visible spectrophotometer. Analysis was conducted in a 100 mm optical cell at a wavelength of 540 nm. Prior to analysis, s-diphenyl carbazide (DPC) in a phosphate medium was added, as recommended by Bartlett (1991). The uv/visible DPC method has some weaknesses. This includes the fact that it is usually utilised in conjunction with acidic extraction of Cr(VI) that does not extract partially water-soluble and water-insoluble Cr(VI) compounds (Ashley et al., 2003). During acidic extraction Cr(VI) is also prone to reduction by Fe(II) (Broadhurst and Maidza, 2006; Pettine and Capri, 2005), which is a commonly-occurring reducing agent for Cr(VI). This was mitigated by extracting Cr(VI) in neutral and pH 9 aqueous environments, prior to the use of the DPC method. Extraction at pH 9 is expected to facilitate the extraction of some of the partially water-soluble Cr(VI) species, while Fe(II) will be oxidised to Fe(III) during neutral or basic extractions. Fe(III) hydroxide precipitates out of solution at these pH levels.
Also, several species, i.e. Cu(II), Fe(III), Hg(II), Mo(VI) and V(V), are known to react with DPC, thus serving as interferences (Ashley et al., 2003). Most of these interferences are not likely to be present. The influence of Fe(III), which might be present, was mitigated as previously discussed.
Although the DPC analytical method is not the most sensitive method (Ashley et al., 2003; Broadhurst and Maidza, 2006; Gómez and Callao, 2006; Pettine and Capri, 2005), it is quick, easy and functional if proper precautions are taken. It is also by far the most commonly used analytical technique for Cr(VI) analyses (Gómez and Calloa, 2006). It has a detection limit of 30 µg·ℓ-1 (Maine et al., 2005), which proved to be more than sufficient for this study.
Results and discussions
Metallurgical-grade ore samples were pulverised for different time periods, i.e. 0.5, 1, 2, 4, 8, 16 min. This procedure was repeated 3 times. The mean and the spread of the obtained d90, d50 and d10 values, as a function of pulverising time, are shown in Fig. 1. d90 is defined as the equivalent particle size for which 90% of the particles are finer. The definition of d50 and d10 can be derived similarly. As expected, the particle sizes became smaller with longer pulverising times. In order to achieve continued size reduction more energy is required, hence the graphs (d90, d50 and d10) reached a plateau with an increase in milling time. It is a well-known fact that the South African chromite ores are friable and break down relatively easily to the crystal grain size (the size of a single chromite spinel crystal). However, to break it down to sizes smaller than this, much more energy would be required (Gu and Wills, 1988).
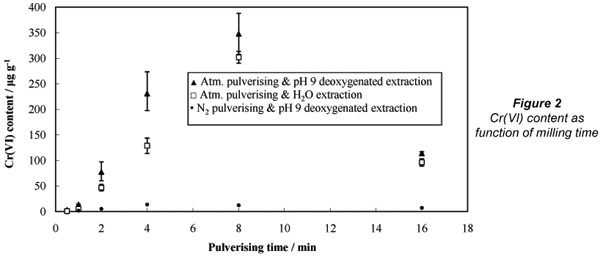
Figure 2 indicates the Cr(VI) content of the above-mentioned chromite samples as a function of pulverising time, with both extraction methods (water and pH 9 de-oxygenated water). As expected, the pH 9 de-oxygenated extraction method extracted more Cr(VI) from the pulverised material than plain water, since it is well-known that alkaline extraction is more effective for quantitative extraction of total Cr(VI) (Ashley et al., 2003; Broadhurst and Maidza, 2006). However, both extraction methods showed the same trends, with Cr(VI) content increasing with increased pulverising time. A maximum amount of Cr(VI) content was reached after 8 min of pulverising. All the samples had an unexpectedly lower Cr(VI) content after 16 min of pulverising – an apparently contradictory phenomenon that will be discussed later.
The most important issue that needs to be clarified for the data is whether the observed Cr(VI) was due to Cr(VI) formation by pulverisation, or Cr(VI) liberation from the ore. In an effort to elucidate the origin of the increased Cr(VI) content with an increase in milling time a very simple alteration to the pulverising method was made. Liquid nitrogen (80 mℓ) was poured into the pulverising container. The lid was placed back on the pulverising container and pressure was kept on the lid by hand. The liquid nitrogen evaporated inside the pulverising container, which resulted in a pressure build-up due to gaseous nitrogen release. This excess pressure was released, while the lid was kept closed with hand-held force to prevent air from re-entering the container. After the pressure build-up had diminished to such an extent that only a small positive N2 pressure remained inside the pulverising container, the container was clamped inside the pulveriser as per normal procedure. This alteration to the procedure resulted in an almost inert nitrogen atmosphere inside the pulverising container. The rubber o-ring between the container and its lid prevented air from re-entering during the subsequent pulverising experiment.
As can be seen from the data presented in Fig. 2, the Cr(VI) content of the chromite samples pulverised in the inert (N2) atmospheres was substantially lower than the chromite pulverised in the normal atmospheres. After 8 min of pulverising, the samples pulverised in the nitrogen-rich atmosphere had at least 2 orders of magnitude lower Cr(VI) content. These data indicate that the Cr(VI) was not present prior to pulverising and that it was not merely liberated by the reduction in size of the chromite particles. It also demonstrated that the much higher Cr(VI) content observed during the normal atmospheric pulverisation was formed due to oxidation.
Beukes and Guest (2001) were the first to indicate that Cr(VI) could be formed during pulverisation of chromium-containing materials. They utilised wet milling (milling in the presence of water) to try to indicate whether Cr(VI) was formed or liberated. This method, however, had some limitations due to the fact that the pulverised material was in contact with water for the entire pulverising period. Cr(VI) could thus have been reduced by metallic iron particles originating from erosion of the container during wet milling. Additionally, Fe(II) could also be liberated from the chromite spinel, which could also reduce Cr(VI). Therefore, dry pulverising in an inert environment is a better method to establish whether Cr(VI) is formed or liberated during pulverising. Aqueous contact of the pulverised material only takes place after pulverising, i.e. during the extraction procedure, and precautions can therefore be taken to minimise the influence of possible Fe(0) and Fe(II) reduction (e.g. by alkaline extraction) (Ashley et al., 2003).
Taking the above-mentioned data and discussions into consideration, it seems apparent that Cr(VI) was formed during normal atmospheric pulverising of chromite. However, the unexpectedly lower Cr(VI) contents after 16 min of pulverising (see Fig. 2) has not been explained thus far. One possible explanation for this phenomenon is the potential liberation of Fe(II) during extended pulverisation. This topic, i.e. liberation of Fe(II) during extended pulverisation of chromite, is currently under investigation and findings will be reported soon. Justification for such a postulation is as follows. The general formula for chromite is (Fe,Mg)O.(Cr,Al,Fe)2O3, with iron in the 2+ and chromium in the 3+ oxidation states (Gu and Wills, 1988). Fe(II) may be partially replaced by Mg(II), Ca(II) and Mn(II), while Cr(III) may be partially replaced by Fe(III), Al(III), Si(IV) and Ti(IV) (Gu and Wills, 1988). Thus, if the spinel crystals are broken, some Fe(II) might be released. Taşdemir (2008) evaluated the grain size distribution of unbroken chromite from 5 different deposits in Turkey and reported mean Ferret particle sizes for the chromite grains to vary between 75.13 and 139.46 µm. Chernet and Marmo (2003) investigated 160 to 250 µm chromite-containing particles recovered during heavy media separation of chromite mined at Kemi in Finland and found that only 0.11% of the chromite grains were smaller than 45 µm. Similar data are not available for South African chromite ores. However, from the experimental particle-size analyses (shown in Fig. 1), it is very clear that at least some chromite crystal grains were broken by the aggressive pulverising technique employed during this study. At 16 min pulverising time the d50 and d10 were 17.77 and 1.93 µm respectively, indicating that a substantial fraction of the particles was likely to be smaller than the possible chromite crystal grain size. Thus it is highly likely that some Fe(II) could have been liberated during the extended pulverising. The liberated Fe(II) could have reduced a fraction of the formed Cr(VI), while the aqueous extractions took place. Although this explanation is speculative, it is the most plausible explanation that can be proposed at present. Research to verify this postulation is currently underway. The actual mechanism of Cr(VI) formation during normal atmospheric pulverisation of chromite was not specifically investigated in this paper and forms part of the afore-mentioned continued research in this field.
Conclusion
From this case study on a metallurgical-grade chromite ore sample, it seems unlikely that South African chromite ores contain Cr(VI). The presence of Cr(VI) in beneficiated South African chromite, as previously reported by Mandiwana et al. (2007), was most likely as a result of the sample preparation technique employed by the afore-mentioned authors. Pulverising of chromite samples in normal atmospheric conditions, prior to chemical analyses, resulted in Cr(VI) formation. Similar over-estimation of the Cr(VI) contents of other chromium-containing materials could be expected if the same technique, i.e. atmospheric pulverising, is applied. Normal atmospheric pulverising of solid samples, prior to chemical analysis for Cr(VI), is therefore not an appropriate sample preparation technique. Inert atmospheric pulverising of solid samples is a viable alternative sample preparation technique, since it does not lead to the formation of Cr(VI). In conclusion, it can be stated that this study has highlighted the need for the development of a standardised solid sample preparation technique, which does not bias the subsequent chemical analysis in any way.
Acknowledgements
The authors wish to thank Xstrata Alloys SA who made this study possible with their financial support. The authors also acknowledge Prof Quentin Campbell and Prof Marthie Coetzee for the use of the particle size analyzer and the pulveriser, respectively.
References
ASHLEY K, HOWE AM, Demange M and Nygren O (2003) Sampling and analysis considerations for the determination of hexavalent chromium in the workplace air. J. Environ. Monit. 5 707-716. [ Links ]
BARTLETT RJ (1991) Chromium cycling in soils and water: links, gaps and methods. Environ. Health Perspect. 92 17-24. [ Links ]
BEUKES JP and GUEST RN (2001) Cr(VI) generation during dry milling. Miner. Eng. 14 423-426. [ Links ]
BROADHURST JL and MAIDZA T (2006) Analytical methods for the determination of chromium in the environment. ICDA (International Chromium Development Association), The chromium file noº15. URL: http://www.icdachromium.com/pdf/publications/CHROMIUM_FILE_N_15.pdf (Accessed 9 September 2009). [ Links ]
CHERNET T and MARMO J (2003) Direct comparison of mechanical and digital size analyses of Kemi chromite, Finland. Miner. Eng. 16 1245-1249. [ Links ]
GÓMEZ V and CALLOA MP (2006) Chromium determination and speciation since 2000. Trends Anal. Chem. 25 1006-1015. [ Links ]
COWEY A (1994) Mining and Metallurgy in South Africa– A Pictorial History. Mintek in association with Phase 4, Randburg, South Africa. [ Links ]
GU F and WILLS BA (1988) Chromite – mineralogy and processing. Miner. Eng. 1 235-240. [ Links ]
HOWAT DD (1994) Chromium in South Africa. J. S. Afr. Inst. Miner. Metall. March 225-240. [ Links ]
IARC (INTERNATIONAL AGENCY FOR RESEARCH ON CANCER) (1997) Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 49: Chromium, Nickel and Welding. URL: http://monographs.iarc.fr/ENG/Monographs/vol49/volume49.pdf (Accessed 9 September 2009). [ Links ]
ICDA (INTERNATIONAL CHROMIUM DEVELOPMENT ASSOCIATION) (2008) Statistical Bulletin – 2008 Edition. 45 pp. [ Links ]
MAINE CF, SMIT JP and GIESEKKE EW (2005) The solid stabilization of soluble wastes generated in the South African ferrochrome industry. WRC Report No. 942/1/05. Water Research Commission, Pretoria. [ Links ]
MANDIWANA KL, PANICHEV N and NGOBENI P (2007) Electrothermal atomic absorption spectrometric determination of Cr(VI) during ferrochrome. J. Hazard. Mater. 145 511-514. [ Links ]
NIAGRU JO (1998) Historical perspectives. In: Niagru JO and Nieboer E (eds.) Chromium in the Nature and Human Environments. Wiley, New York. 1-9. [ Links ]
PETTINE M and CAPRI S (2005) Digestion treatments and risks of Cr(III)-Cr(VI) interconversions during Cr(VI) determination in soils and sediments – a review. Anal. Chim. Acta 540 231-238. [ Links ]
RIEKKOAL-VANHANEN M (1999) Finnish Expert Report on Best Available Techniques in Ferrochromium Production. The Finnish Environment 314. Finnish Environment Institute, Helsinki. 49 pp. [ Links ]
TAŞDEMIR A (2008) Evaluation of grain size distribution of unbroken chromites. Miner. Eng. 21 711-719. [ Links ]
Received 15 May 2009; accepted in revised form 12 November 2009.
* To whom all correspondence should be addressed.
+2718 299 2337; fax: +2718 299 2350;
e-mail: paul.beukes@nwu.ac.za


{kind=link}
{kind=link}