










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.35 n.3 Pretoria Apr. 2009
Removal of copper and cobalt from aqueous solutions using natural clinoptilolite
BB MambaI, *; DW NyembeI; AF Mulaba-BafubiandiII
IDepartment of Chemical Technology, Faculty of Science, University of Johannesburg, PO Box 17011, Doornfontein, South Africa
IIMinerals Processing and Technology, Department of Extraction Metallurgy, Faculty of Engineering and the Built Environment, University of Johannesburg, PO Box 526, Wits 2050, South Africa
ABSTRACT
Southern African clinoptilolite capability as an ion-exchanger with respect to Cu2+ and Co2+ was investigated in order to consider its possible application at removing metals from aqueous solutions. The column method was used in the cation-exchange processes with synthetic solution concentrations of 0.07 M (3.86 g/ℓ), 0.33 M (19.31 g/ℓ) and 0.66 M (38.63 g/ℓ) of Cu2+ solution and 0.07 M (3.34 g/ℓ), 0.33 M (16.69 g/ℓ) and 0.66 M (33.37 g/ℓ) of Co2+ solution. Synthetic non-mixed sulphate solutions of copper and cobalt recorded maximum cation uptakes of 79% and 63% with 0.02 M HCl-activated clinoptilolite respectively. From the Cu/Co mixed solutions, both cobalt and copper recorded a 79% uptake with 0.02 M HCl-activation. The 0.04 M HCl activation gave percentage removals of 79% and 77% for Co2+ and Cu2+ respectively. In the ion-exchange evaluation part of the study, it was found that in every concentration range, the adsorption mass ratio of clinoptilolite to metal concentration conformed to both Langmuir and Freundlich adsorption isotherms. However, the non-mixed aqueous solutions of Cu2+ and Co2+ fitted mainly the Langmuir equation. It was found that the adsorption process depends on the hydrated radius of the cation being exchanged, the concentration of the acid that activates the clinoptilolite and the concentration of the targeted cation in solution.
Keywords: clinoptilolite, cation exchange, copper, cobalt; wastewater
Introduction
Water pollutants emanating from metallurgical operations have been highlighted as one of the avenues by which groundwater can be polluted thus threatening valuable water sources. The need for clean drinking water is vital for the promotion of good health and with it comes the challenge to provide safe and high-quality drinking water. Groundwater is one of the major sources of drinking water in South Africa. These freshwater sources are faced with the challenge of deteriorating quality due to the prevalence of pollution. The need for removal of metal species in water and the general purification of these waters cannot be overemphasised. A number of purification methods are currently employed in the water purification industry and these include ion exchange, ultrafiltration, phytoextraction, electrodialysis and reverse osmosis (Geselbracht, 1996; Schnoor, 1997). However, industrial wastewaters often contain a cuisine of metals that may impose cost implications when it comes to purification.
Among the water purification technologies, ion exchange is most commonly employed in industrial water treatment (Inglezakis et al., 2005). The ion-exchange reaction occurs between two or more phases, one of which is liquid (solution or melt), whereby an exchange occurs of two or more ions that are more or less strongly bound to each phase. The ion exchanger may be a resin, activated carbon, a zeolite or clay mineral. Zeolites, synthetic or natural, are aluminosilicates that have a framework structure with enclosed cavities and tunnels which are occupied by freely moving hydrated cations that make ion exchange possible.
Clinoptilolite is the most abundant and cosmopolitan zeolite (Inglezakis et al., 2005) and it has been widely exploited for its ion-exchange capabilities since it can easily exchange its interstitial sodium for external cations in solution (Kuronen et al., 2006). Natural zeolites often do not perform optimally as required for industrial applications. Therefore, chemical conditioning of zeolites to improve their performance is often carried out. This conditioning removes certain cations from zeolites that may hinder ion exchange and expose the easily exchangeable ones. It has been previously reported in the literature (Zamzow et al., 1992) that sodium-type natural clinoptilolite favours the exchange of Cu2+ over Co2+. Cation-exchange isotherms of Cu2+ exchange on chabazite and clinoptilolite (Semmens and Seyfarth, 1978; Blanchard et al., 1984; Assenov et al., 1988), Co2+ exchange on clinoptilolite (Blanchard et al., 1984), Zn-exchange on clinoptilolite (Semmens and Seyfarth 1978; Blanchard et al., 1984; Assenov et al. 1988) and Cr3+ exchange on phillipsite and chabazite (Pansini et al., 1991) are also reported in literature.
Herein, we report on the ability of the South African natural and modified clinoptilolite at removing heavy metals such as Cu and Co from their synthetic solution using the ion-exchange capability of the zeolite. The effect of concentration of the aqueous solutions and the degree of HCl activation of the clinoptilolite in its removal efficiency of the clinoptilolite were also studied.
Experimental
Zeolite source, characterisation and conditioning
The natural clinoptilolite used in this study was sourced from the Vulture Creek, KwaZulu-Natal Province of South Africa. In preparation for mineralogical studies and characterisation, the clinoptilolite was crushed and milled into powder with average particle sizes of approximately 75 µm. The powder was then examined using an X-ray powder diffractometer (XRD) Phillips X'pert Model 0993 to determine its composition. Its elemental composition was determined using X-ray fluorescence spectroscopy (XRF, Phillips Magix Pro) while the surface area was analyzed using BET (Tristar 3000). The measurements were done under a nitrogen atmosphere. Prior to porosity and surface area analysis, 2 g of sample was first degassed and nitrogen gas was flushed through for 4 h at 120ºC. Clinoptilolite grains of sizes in the range of 2.8 mm to 5.6 mm were used for adsorption studies. A fraction of these grains was treated in HCl at concentrations of 0.02 M and 0.04 M at room temperature over a period of 8 h. The clinoptilolite was then washed in deionised water to remove the fine fractions and thereafter dried in the oven at 50ºC for 24 h.
Synthetic solutions
Solutions of Cu and Co were prepared by dissolving CoSO4.7H2O and CuSO4.5H2O in deionised water at pH 6.5. The non-mixed Cu2+ and Co2+ aqueous solutions of concentrations 0.0020 M, 0.0698 M and 0.2000 M which corresponded to 0.032 g, 1.109 g and 3.177 g of Cu2+ and 0.030 g, 1.028 g and 2.947 g of Co2+ were prepared by dissolving CoSO4.7H2O and CuSO4.5H2O salts in deionised water in 250 mℓ volumetric flasks.
Studies on the mixed solutions of copper and cobalt were done with solutions of Cu and Co prepared at stoichiometric ratios of Co:Cu – 1:1, 1:5, 1:9, 5:1 and 9:5 which corresponded to these concentrations of Co:Cu - 0.0020:0.0020 g/ℓ, 0.0020:0.0698 g/ℓ, 0.0020:0.2000 g/ℓ, 0.0698:0.0020 g/ℓ and 0.2000:0.0020 g/ℓ respectively. Studies on the Co/Cu mixed synthetic solutions were done with solutions of copper and cobalt prepared at stoichiometric ratios of Co:Cu – 1:1, 1:5, 1:9, 5:1 and 9:1 which corresponded to these concentrations of Co:Cu – 0.0020:0.0020 g/ℓ, 0.0020:0.0698 g/ℓ, 0.0020:0.2000 g/ℓ, 0.0698:0.0020 g/ℓ and 0.2000:0.0020 g/ℓ respectively. These solutions were assayed using atomic absorption spectroscopy (AAS), (model Varian Spectra (20/20)).
Batch adsorption studies on synthetic solutions
The Cu and Co ion-exchange processes on the clinoptilolite were conducted at room temperature. Glass columns of 2 cm diameter and 30 cm of length were pre-loaded with 25 g of either natural clinoptilolite (as received) or HCl-activated clinoptilolite. Aliquots of 25 mℓ of the prepared Cu- and Co-bearing solutions of desired concentrations were passed through each of the two types of zeolites. These were afforded the same solution-zeolite contact time. After passing through the zeolite-packed column the resultant solutions were assayed using atomic absorption spectroscopy (AAS) in order to ascertain the zeolite's removal efficiency. The flame type used was air-acetylene and the adsorption wavelengths for the two metals were Cu (324.7 nm) and Co (240.7 nm). Standards of 1 000 (mg/ℓ), 2 000 (mg/ℓ) 3 000 (mg/ℓ) were then prepared and a calibration curve was drawn using these standards. Dilution was applied stoichiometrically where the concentrations of the unknown solutions of copper and cobalt exceeded the standards' concentration range of the standards.
The metal-removal percentage from the solutions and distribution ratio were calculated as follows:
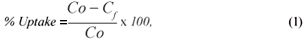
where:
Co and Cf are the initial and final metal concentrations, respectively.
Results and discussion
Clinoptilolite characterisation
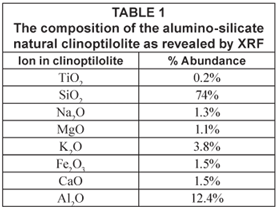
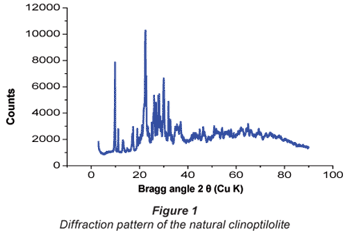
Results of the X-ray fluorescence (XRF) analysis are presented in Table 1. This characterisation showed that clinoptilolite contained exchangeable ions of sodium, potassium, calcium and magnesium. This zeolite has an Si/Al ratio of 5.96 (mol/mol) and the corresponding ratio of (Na + K)/Ca was 3.4. XRF also confirmed that the zeolite was a high silica clinoptilolite enriched with Mg, K and Na. The XRD plot in Fig. 1 shows a typical mineralogical diffraction pattern of a crystallite with a composition of 70% SiO2, 12% Al2O3, 2% Na2O, 5% K2O, 2% CaO and 2.5% Fe2O3.
Metal removal from synthetic solutions
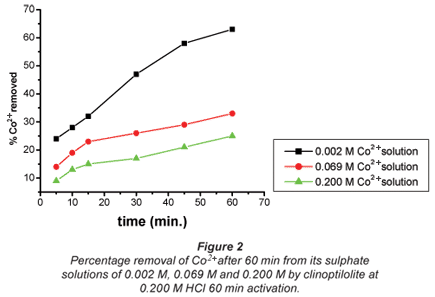
The ion-exchange kinetics of Co2+ and Cu2+ as a function of their concentrations in their respective solutions were studied at room temperature by varying their concentrations with time and keeping all other parameters constant. From the results shown in Figs. 2 and 3, it can be confirmed that the percentage metal removal decreased with an increase in metal concentration in the aqueous solutions. This may be due to the fact that particles always diffuse faster when they are less packed, thus they move faster in dilute solutions and more slowly in concentrated solutions.
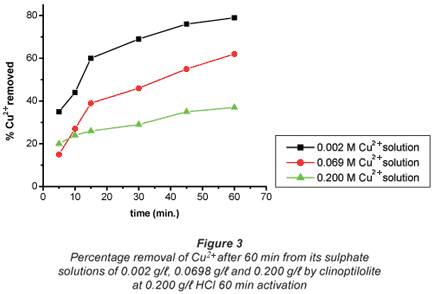
Metal removal from the double-component systems
The copper/cobalt mixed solutions exhibited results similar to the single-salt solutions in the sense that the removal efficiency was high with dilute solutions than it was with concentrated solutions and the results shown in Figs. 4 to 7 confirm this assertion. High uptakes ranging between 70 to 79% were recorded with both Cu and Co removal in the concentration ratios of 0.07:0.07 M.
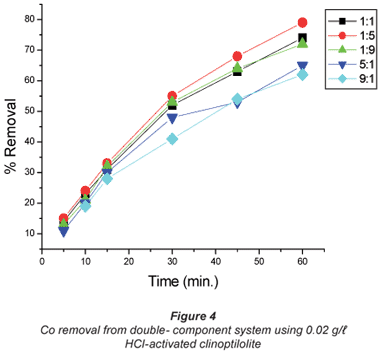

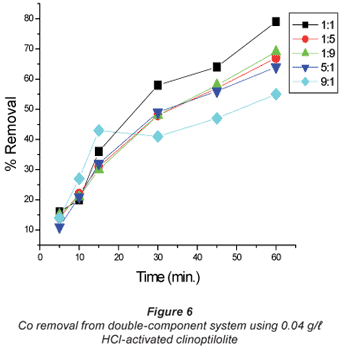
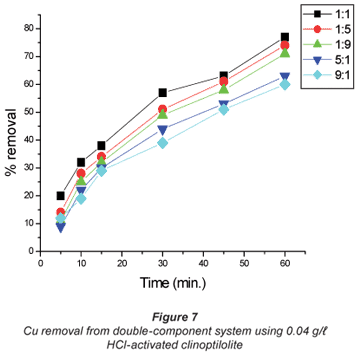
In solutions where there was more cobalt than copper in terms of the ratio (Co:Cu = 9:1 and 5:1) there seemed to be difficulty in removing either of the metals using the 0.04 M HCl- activated clinoptilolite. Co removal efficiency in Co:Cu = 9:1 solutions using 0.02 M HCl was 62% and was reduced to 48% when 0.04 M HCl-activated clinoptilolite was used while a ratio of Co:Cu = 5:1 showed a decrease in removal efficiency from 64% using 0.02 M HCl-activated clinoptilolite to 57% when 0.04 M HCl-activated clinoptilolite was used. On the other hand, Cu removal from the Co: Cu = 9:1 ratio showed a removal efficiency decrease from 59% when 0.02 M HCl was used to 55% when 0.04 M HCl-activated clinoptilolite was used. It is possible that there is an intermediate complex that is formed between Co2+, Cu2+ and water molecules that is too bulky to favour fast removal of these metals from the aqueous solution. The presence of Cu2+ in high concentrations (Co:Cu = 1:9 and 1:5) in the double-component solution favours the removal of both Co and Cu. This somehow contradicts the notion that less concentrated metal solutions favour fast removal of that specific metal than concentrated solutions. However, the fact that the highest removal efficiencies with these solutions are recorded with the more dilute solutions agrees with the notion.
Langmuir and Freundlich adsorption isotherms
The sorption data was subjected to Langmuir and Freundlich adsorption isotherms. The equilibrium data for single metal solutions was correlated with the Langmuir isotherm according the following equation (Langmuir, 1918):

where:
Cr is the equilibrium concentration of the metal in solution remaining after adsorption (mg/ℓ)
x/m is the number of metal ions adsorbed onto the clinoptilolite
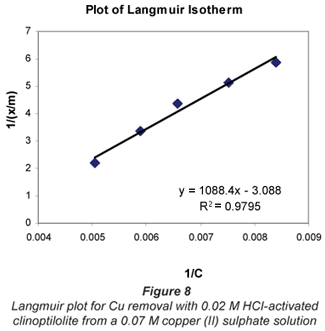
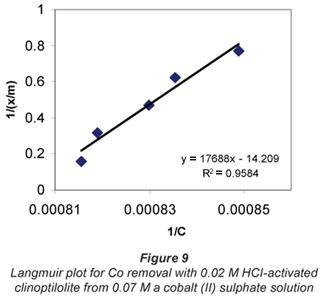
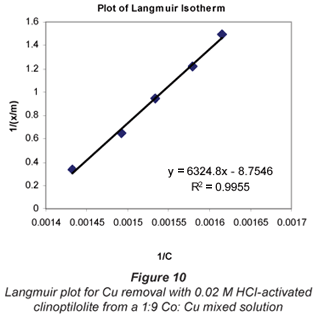
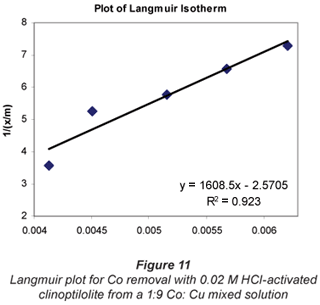
a and b are Langmuir constants related to adsorption capacity and adsorption energy, respectively. Constant a is the maximum sorption capacity and represents single layer coverage of the adsorbent (clinoptilolite) in contact with the adsorbate (metal solution). The variable b represents enthalpy of adsorption and changes with temperature. Application of the Langmuir Model to the data is shown in Figs. 8 to 11.
The Langmuir equation can be rearranged in a linear equation form and rewritten as follows:
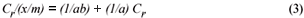
To make the Langmuir plots, Cr/(x/m) is plotted against Cr and a straight-line graph was obtained for this plot. The data of the non-mixed salt solutions fit the Langmuir Model very well with R2 values > 0.9 for the dilute sulphate solutions of both Cu and Co. According to the Langmuir Model and R2 correlation values, Cu2+ uptake is more favoured than Co2+.
The ion-exchange capacity of Cu2+ and Co2+ indicates that clinoptilolite favours Cu2+ over Co2+ ions. In solution, these ions are present in aqua-ionic forms and Cu2+ is found in a tetra-aqua form while Co2+ is exists in the hexa-aqua form. This suggests that Cu2+ is surrounded by four molecules of water while Co2+ is surrounded by six. These hydrated ions pass through the clinoptilolite (Jama and Yucell, 1990) and although the charges of the ions are the same, they have different hydrated radii – Cu2+ has a lower hydrated radius than Co2+. The adsorption of a metal ion by an ion-exchanger largely depends on its hydrated radius. The larger the diameter the slower its mobility and the less likely its exchange would occur (Erdem et al., 2004). It appears that for easy ion exchange some of the water molecules surrounding the cation must be lost in order to initiate the ion-exchange process.
The Langmuir Model effectively describes the adsorption data obtained in these experiments with all values of R2 being greater than 0.99 for the dilute solutions of 0.07 M metal sulphate being exchanged with 0.02 M HCl-activated clinoptilolite. The isotherms also confirmed the selectivity of clinoptilolite as being more favourable for Cu2+ than Co2+. The clinoptilolite was observed to generally have good selectivity for both cations. This could be attributed to the high Si/Al ratio of clinoptilolite with a low ionic field that results in good selectivity.
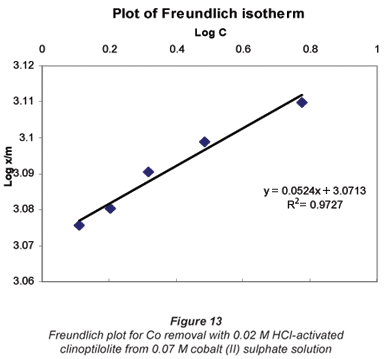
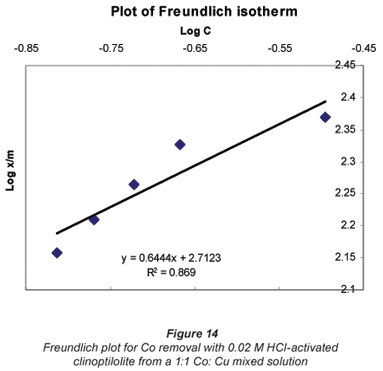
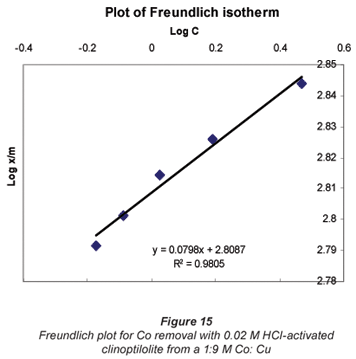
Another mathematical model used to fit the data obtained in this study was the Freundlich isotherm (Altin et al., 1998). This model is known for its good fit of data over a wide range of concentrations. It gives an equation that includes the heterogeneity of the surface of the ion exchanger and the exponential distribution of active sites and their energies. The Freundlich equation is given by:
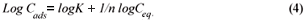
where:
Ceq is the equilibrium concentration in ppm
Cads is the number of metal ions adsorbed onto the clinoptilolite
K is related to temperature
n is a characteristic constant for any system under study
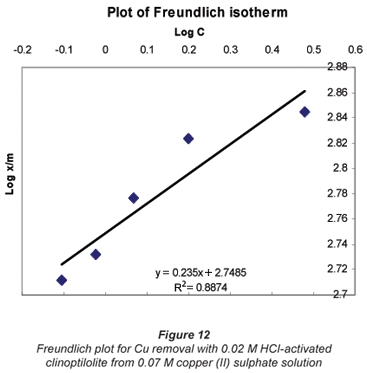
It was observed that the data followed the Freundlich Model as well. The R2 values were generally higher than 0.95.
The selectivity of a zeolite for one ion over another in a matrix is a result of physicochemical and stereochemical factors. These are hydrated radii, the hydration enthalpy of the cation and the space requirements in the micropores of clinoptilolite (Inglezakis et al., 2005). Every metal in solution possesses a hydrated layer which has a characteristic thickness and degree of stability. It has been proven that ions with large radii often show large rejections while smaller ions are often favoured by exchangers (Tansel et al., 2006). Cu2+ is a smaller ion with a smaller hydrated radius than Co2+ and therefore Cu2+ is highly favoured for removal by clinoptilolite within the parameters of this study.
It is also known that the hydration enthalpy of Cu2+ is low due to the number of water molecules solvating it (Carrillo-Tripp et al., 2006). The easy removal of Cu compared to Co can be attributed to the geometry of the hydrated copper ion (Inglezakis et al., 2005). Cu2+ forms a square-planar complex in water, [Cu (H2O)4]2+, while Co2+ forms a tetrahedral aqua-complex, [Co(H2O)6]2+ due to its hexavalent ion. The square planar complex is less bulky and therefore migrates easily in solution compared to the bulky tetrahedral aqua-complex
The strength of the exchange sites on the zeolite also affects the exchange rate. Natural clinoptilolite is weakly acidic hence the rate of ion exchange in clinoptilolite is expected to be high. This is even more so with the acid-activated clinoptilolite.
Effect of solution concentration
In general, natural clinoptilolite appeared to remove the metals more efficiently from dilute solutions than from concentrated ones. Athanasiadis and Helmreich (2004) reported that high metal-solution concentrations showed low ion-exchange rates compared to low concentrations. These researchers concluded that this was due to the high concentration of the counter ion in the solution which was generally true for all the analytes. However, an outlier was observed with Co removal from the Co mixed solutions using 0.04 M HCl-activated clinoptilolite (Fig. 6). This must have been due to an unevenly mixed size proportion of the zeolite used. It is possible that more of the smaller size range of particles was present in this batch. Small particle size increases the exchange efficiency due to surface area: volume ratio. The cation being exchanged moves a shorter distance in smaller grains than in larger ones thus speeding the exchange rate.
Effect of HCl conditioning
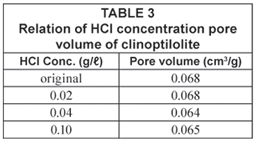
The BET analysis shows no significant changes in pore volume with increase in HCl activation concentration of the zeolites. Analyses of original, 0.02 M, 0.04 M and 0.10 M HCl-activated clinoptilolite revealed that there is a decrease pore volume and surface area as the HCl concentration increases. The results of pore volume measurements are shown in Table 3.
The SEM image in Fig. 16 shows a more open structure of the HCl-activated clinoptilolite's surface compared to the inactivated zeolite in Fig. 17. This could explain why acid-activated clinoptilolite performed better than the original zeolite. There were some morphological changes brought about by acid activation on clinoptilolite structure when comparing the two SEM images.


The FTIR spectra in Fig. 18 give a clear picture about the effect of chemical conditioning. Acid treatment is said to remove the non-zeolitic components thus increasing the concentration of zeolite minerals. Elimination of this material requires more complex procedures (Vanloon and Duffy, 2000; Al-Degs et al., 2004). Infrared techniques have been used by previous researchers for the identification of soil and clay minerals (Gadsden, 1975). Figure 18 depicts the FTIR-spectra of the original and acid-treated forms of the clinoptilolite used in this study.
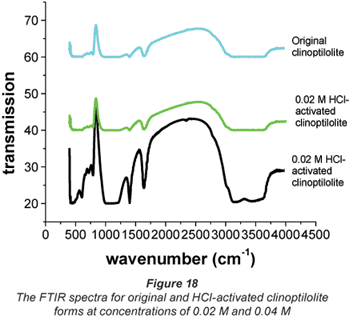
The results obtained from FTIR spectroscopy agree with the observations made with respect to the performance of the clinoptilolite where 0.02 M HCl-activated zeolite was the best ion-exchanger, i.e. higher percentage extractions were obtained with this form of zeolite than those obtained with original and 0.04 M HCl activation concentration. Over the range of 4 000 and 3 600·cm-1 the original and 0.04 M HCl-activated forms of clinoptilolite showed no peaks while the 0.02 M HCl-activated form showed peaks. Peaks observed at this range are typical of water absorption (Majedova, 2003; Al-Degs et al., 2004). This shows that water adsorption and retention by clinoptilolite is increased by HCl activation at 0.02 M concentration and can be used to explain the good performance of this form of activated clinoptilolite in ion exchange compared to the other 2 forms (original and 0.04 M HCl-activated forms).
Over the range of 2 500 and 2 000·cm-1, the 0.02 M HCl-activated clinoptilolite showed 2 intensive peaks and yet again the original and the 0.04 M HCl-activated forms showed none. This could be as a result of 0.02 M HCl activation washing out the non- zeolitic impurities present in the original form as confirmed by XRD, XRF and SEM-EDS (Gadsden, 1975). The disappearance of these peaks with the 0.04 M HCl-activated form could be due to the strength of the acid resulting in the destruction of the active sites observed with the 0.02 M HCl-activated clinoptilolite. There were broad peaks observed for all the clinoptilolite forms at 1 558·cm-1 which may be due to the bending vibrations of adsorbed water. This is expected since given its porous structure, desiccation of the zeolite at high temperatures 50°C will increase its water absorption properties (Ng and Mintova, 2008). Zeolites that have K+ in high amounts have low water-adsorption capacity (Hunger et al. 1999; Kirchhock et al. 2000). This could probably explain why the original clinoptilolite does not show these peaks. It is also possible that the K+ in the acid activated zeolite was leached out by the acid, thus increasing their water sorption capacity.
The identification of peaks between 1 500 and 1 000·cm-1 FTIR indicates the presence of a high content of calcite in the sample as confirmed by SEM-EDS. The strong band at 1 341· cm-1 (due to Si–O stretching) is the main characteristic band for quartz (Al-Degs et al., 2004). The peaks observed between 1 000 and 600·cm-1 are present in all the forms of clinoptilolite, one characteristic band appears at 836·cm-1 for all the forms. This is the quartz band (Al-Degs et al., 2004). The peak that appears at 753·cm-1 for the original form of clinoptilolite appears at 759·cm-1 for the activated zeolite. There is a peak that appears at 686·cm-1 for the original form while for the acid-activated form it appears at 635·cm-1. This shift could be attributed to the action of the acid.
In the non-mixed salt solutions, activated clinoptilolite generally performed better at taking up metal than the original zeolite. The original clinoptilolite showed capacity to remove 37% Cu and 39% Co. Uptakes of 79% and 63% for Cu2+ and Co2+ were recorded with 0.02 M and 0.04M HCl activation, respectively. In the mixed solutions, both cobalt and copper recorded 79% uptake with 0.02 M HCl activation. The 0.04 M HCl-activated zeolite gave percentage removals of 79% and 77% for Co and Cu respectively.
It has been documented that acid treatment of natural clinoptilolite improves its adsorbent properties (Korkuna et al., 2006; Inglezakiz et al., 2001; Athanasiadis et al., 2005). This is due to de-catination, de-alumination and the dissolution of amorphous silica fragments blocking the channels. A study by Korkuna et al. (2006) also revealed that there is a change in the clinoptilolite structure after acid treatment with dilute acid activations accounting for improved removal capacity of the clinoptilolites. Studies by Vasylechko et al. (1999) and Hernandez (2000) confirm findings in this study with respect to the effect of acid conditioning of natural clinoptilolite.
Conclusions
Relatively speaking, it was easier to remove Cu2+ than Co2+ from the non-mixed solutions. Uptakes of 79% for Cu2+ and 63% for Co2+ were obtained. The removal efficiency of the metals by the zeolite from dilute solutions was higher than from concentrated solutions for both non-mixed and mixed solutions. Clinoptilolite activated with 0.02M HCl was the best performing zeolite for both Co2+ and Cu2+ removal in non-mixed solutions. Uptakes of 79% and 63% for Cu2+ and Co2+ were recorded with 0.02 M and 0.04M HCl activation, respectively.
Acknowledgements
The authors would like to acknowledge financial support from the University of Johannesburg, Eskom's Tertiary Education Support Programme (TESP) and the National Research Foundation (NRF). The authors also wish to thank Pratley (South Africa) for providing the natural clinoptilolite samples that were used in this study.
References
AL-DEGS S, EL-BARGHOUTHIA M, ISSAA A, KHRAISHEHB M and WALKER G (2006) Sorption of Zn(II), Pb(II) and Co(II) using natural sorbents: equilibrium and kinetic studies. Water Res. 40 2645-2658. [ Links ]
AL-DEGS Y, TUTUNJI M and BAKER H (2003) Isothermal and Kinetic Sorption Behaviour of Pb2+ ions on natural silicate minerals. J. Clay Miner. 38 501-509. [ Links ]
ALTIN O, ÖZBELGE H and DOGU T (1998) Use of general purpose adsorption isotherms for heavy metal-clay mineral interactions. J. Colloid and Interface Sci. 198 130-140. [ Links ]
ATHANASIADIS and, HELMREICH B (2005) Influence of chemical conditioning on the ion-exchange capacity and on kinetic of zinc uptake by clinoptilolite. Water Res. 39 1527-1532. [ Links ]
ATHANASIADIS K, HELMREICH B, PETER C, HILLIGES R and WILDERER P (2004) On-site treatment of runoff from roofs and prior to infiltration. Water and Environmental Management Series. IWA Publishing, London, UK. 61-68. [ Links ]
BARRER R M (1950) Ion-exchange and ion-sieve processes in crystalline zeolites. J. Chem. Soc. London 2342-2350. [ Links ]
BLANCHARD G, MAUNAYE M and MARTIN G (1984) Removal of heavy metals from waters by means of natural zeolites. Water Res. 18 (12) 1501-1507. [ Links ]
BRIGATTI M, FRANCHINI G, FRIGIERI P, GARDINALI C MEDICI L and POPPI L (1999) Treatment of industrial wastewaters using zeolite and sepiolite. Natural Microporous Mater. 77 163-168. [ Links ]
CARRILO-TRIPP M, SAN-ROMAN M L HERNANDEZ and COBOS J (2006) Ion hydration in nanopores and the molecular basis of selectivity. Biophys. Chem. 124 243-250. [ Links ]
EDERM E, KARAPINAR N and DONAT R (2004)The removal of heavy metal cations by natural zeolites. J. Colloid and Interface Sci. 280 309-314. [ Links ]
HAJJAJI M, KACIM S, ALAM A, EL-BOUADILI A and EL-MOUNTASSIR M (2001) Chemical and mineralogical characterization of clay taken from the moroccan meseta and a study of the interaction between its fine fraction and methylene blue. Appl. Clay Sci. 20 1-12. [ Links ]
GISELBARCHT J (1996) Microfiltration/reverse osmosis pilot trials for Livermore, California, advanced water reclamation. Water Reuse Proceedings: AWWA 187. [ Links ]
HERNANDEZ M A (2000) Nitrogen-sorption characteristics of the microporous structure of clinoptilolite-type zeolites. J. Porous Mater. 7 (4) 443-454. [ Links ]
INGLEZAKIS V J and GRIGOROPOULOU H (2004) Effects of operating conditions on the removal of heavy metals by zeolite in fixed bed reactors. J. Hazardous Mater. B112 37-43. [ Links ]
INGLEZAKIS V J, ZORPAS A A, LOIZIDOU M D AND GRIGOLOPOULOU H P (2005) The effect of competitive cations and anions on ion exchange of heavy metals. Sep. Purif. Technol. 46 202-207. [ Links ]
JAMA M A and YUCEL H (1990) Equilibrium studies of sodium-ammonium, potassium-ammonium and calcium-ammonium exchanges on clinoptilolite zeolite. Sep. Sci. Technol. 24 (15) 1393-1416. [ Links ]
KURONEN M, WELLER M, TOWNSEND R and HARJULA R (2006) Ion exchange selectivity and structural changes in highly aluminous zeolite. React. Funct. Pol.66 1350-1361. [ Links ]
LANGMUIR I (1918) The adsorption of gases on plane surfaces of glass, micra and platinum. Am. Chem. Soc. 40 1361-1402. [ Links ]
KIRSCHHOCK C, HUNGER B, MARTENS J and JACOBS P A (2000) Localisation of residual water in alkali-metal cation-exchanged x and y type zeolites. J. Phys. Chem. B 104 439-448. [ Links ]
MADEJOVA J (2003) FTIR techniques in clay minerals studies: A review. Vib. Spectrosc. 31 1-10. [ Links ]
NG E-P, DELMOTTE L and MINTOVA S (2008) Environmentally benign synthesis of nanosized aluminophosphate enhanced by microwave heating. Green Chem. 10 1043-1048. [ Links ]
NG E and MINTOVA S (2008) Nanoporous materials with enhanced hydrophilicity and high water sorption capacity. Microporous and Mesoporous Materials 114 1-26. [ Links ]
SEMMENS M, SEYFARTH IN, SAND LB and MUMPTON FA (1978) (eds.) (1996) Natural Zeolites. Occurrence, Properties and Use. Pergamon Press. Elmsford, NY. 517 pp. [ Links ]
SCHNOOR J L (1997) Ground water remediation technologies analysis centre, Pittsburgh Phytoremediation TE 97-01. [ Links ]
OZKAR S and TUNALI NK (1997) Inorganic Chemistry. Gazi University Publication, Ankara, Turkey. 136 pp. [ Links ]
PANSINI M, COLELLA C and DE'GENNARO M (1991) Chromium removal from water by ion exchange using zeolite. Desalination 83 145-157. [ Links ]
VASYLECHKO VO, GRYSHCHOUK GV, LEBEDYNETS LO, LEBODA R, and SKUBISZEWSKA-ZIEBA J (1999) Investigation of usefulness of Transcarpathian zeolites in trace analysis of waters. Application of mordenite for the preconcentration of trace amounts of copper and cadmium. Chem. Anal. (Warsaw) 44 (6) 1013-1024. [ Links ]
ZAMZOW MJ and MURPHY JE (1992) Removal of metal cations from water using zeolites. Sep. Sci. Technol. 27 1969-1984. [ Links ]
Received 12 November 2008; accepted in revised form 16 March 2009.
* To whom all correspondence should be addressed.
+2711 559 6516; fax: +2711 559 6425;
e-mail: bmamba@uj.ac.za

