










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.35 n.3 Pretoria Apr. 2009
Sequential injection spectrophotometric determination of V(V) in environmental polluted waters
Eliana S Silva; Paula CAG Pinto*; José LFC Lima; M Lúcia MFS Saraiva
Requimte, Serviço de Química-Física, Faculdade de Farmácia, Universidade do Porto, Rua Aníbal Cunha, 164, 4099-030 Porto, Portugal
ABSTRACT
A fast and robust sequential injection analysis (SIA) methodology for routine determination of V(V) in environmental polluted waters is presented. The determination was based on the oxidation of dopamine (DP) by V(V) in acidic medium followed by coupling of the formed intermediary with 4-aminoantipyrine (4AAP) to yield a violet product (λ=565 nm).
The operation mode of the SIA system allowed the implementation of a stopped flow procedure in which the reaction zone was stopped for 180 s in the reaction coil before reaching the spectrophotometric detector, with the aim of increasing the sensitivity.
Linear calibration plots were obtained for V(V) concentrations between 0.50 and 5.0 mg·ℓ-1. The developed methodology exhibits a good precision, with relative standard deviation (rsd) < 2.0% (n=15) and the detection limit was 0.39 mg·ℓ-1.
The presented SIA methodology was applied to the determination of V(V) in 10 water samples and in a wastewater reference certified sample.
The determination of V(V) by the developed automatic procedure involved the consumption of 1.9 mg of 4AAP and 2.9 mg of DP and the production of 2.35 mℓ of effluent.
Keywords: sequential injection analysis (SIA), vanadium (V), environmental polluted water, spectrophotometric
Introduction
Vanadium is a metallic element widely distributed in nature that in vestigial amounts is an essential element for cell growth, and is also important for the prevention of some heart diseases. Regarding its importance for plants and microorganisms, vanadium is a growth factor, being involved in nitrogen fixation and accumulation (Mukherjee et al., 2004). In large amounts vanadium can become a relatively toxic element, depending on its oxidation state, so that in aqueous environments vanadium (V) (V(V)) is more toxic than vanadium(IV) (V(IV)). Some cases of vanadium poisoning have been related to nervous depression, coughing, vomiting, diarrhoea, anaemia and increased risk of lung cancer. Bronchitis and bronchopneumonia as well as irritation of eyes and skin can occur due to occupational exposure to airborne vanadium (Klaassen, 2008).
The utilisation of vanadium and its compounds in steel and petrochemical industries has resulted in the release of large amounts of vanadium compounds to air, water and soil, so that the control of vanadium content, mainly V(V), in waste waters and environmental samples from highly polluted areas became an important issue (Pyrzyńska and Wierzbicki, 2004).
The determination of V(V) in wastewaters has been performed by spectrophotometry (Abbaspour and Mirzajani, 2006; Deb et al., 1991; Kamavisdar and Patel, 2000; Mohamed and Fawy, 2000), electrothermal atomic absorption spectrometry (emberyová et al., 2007) and ICP-MS (Yeh and Jiang, 2004). These methodologies involve either complicated and laborious procedures or expensive equipments, being unsuitable for routine application. The application of spectrophotometric methodologies for the determination of V(V) in aqueous matrixes has been reviewed twice in the past 2 decades (Taylor and Van Staden, 1994; Pyrzyńska, 2005). Most of these methodologies are based on the formation of complexes and redox or catalytic reactions. It is also possible to find recent works involving the speciation and determination of vanadium in environmental samples (Chen and Owens, 2008). Even though these techniques can represent sensitive alternatives for the determination of V(V) they are time-consuming and must be performed batch-wise, requiring the constant presence of a trained operator.
To overcome some of these drawbacks some flow injection analysis (FIA) methodologies have been proposed for the determination of V(V) in environmental water samples. Some of these methodologies involved the speciation (Yamane et al., 1998) and/or pre-concentration (Matsuoka et al., 1995; Yang et al., 1996; Wuilloud et al., 2001; Moyano et al., 2006) of V(V) and V(IV), but most of them are spectrophotometric procedures based on reactions in which V(V) is able to catalyse the oxidation of several compounds (Nakano et al., 1989; Ensafi and Kazemzadeh, 1996; Shiobara et al., 1999; Nakano et al., 2002). A FIA procedure based on chemiluminometric detection can also be found in the literature (Li et al., 2002). These analytical techniques represent a step forward in the automation of the determination of V(V), presenting advantages in terms of time consumption. However, some of these procedures require expensive equipments (Wuilloud et al., 2001; Yang et al., 1996) and most of them still exhibit a high dependence on the operator. Furthermore, due to their continuous mode of operation, FIA techniques in general involve significant consumptions of reagents solutions and production of effluents so that it is important to research new automatic methodologies.
In an attempt to provide a fast and automatic method for the routine analysis of V(V) in wastewaters and highly polluted environmental samples the determination was implemented in a sequential injection analysis (SIA) system, in agreement with the actual concerns of the green chemistry. SIA (Ruzicka and Marshall, 1990) is an automated analytical method developed to address some of the limitations of FIA. It makes use of a simpler, more robust single channel manifold whose versatility is centred around a selection valve. Each port of this valve is dedicated to a specific purpose and the combinations of sample, standards, reagents and detectors around the valve are easily modified to suit a particular analysis. The computer control mode of operation of the systems allows the aspiration of precise volumes and an effective utilisation of the solutions, minimising the consumption of sample and reagent solutions and the generation of effluents (Lenehan et al., 2002). Moreover, the portable nature of the developed systems and the possibility of performing real-time analysis make SIA a very interesting tool for environmental water pollution monitoring, with potential to perform multi-parametric determinations.
The sequential injection determination of V(V) was based on the oxidation of dopamine (DP) by V(V) in acidic medium followed by coupling of the formed intermediary with 4-aminoantipyrine (4AAP) to yield a violet product (λ=565 nm).
The implementation of the spectrophotometric determination of V(V) in a SIA system is intended to provide an automatic method suitable for routine analysis and environmental in situ monitoring that could be an alternative to the existing procedures used for the same purpose.
Experimental
Reagents
All solutions were prepared using chemicals of analytical reagent grade and high purity water (milli-Q), with a specific conductance <0.1 µS·cm-1.
4-Aminoantypirine (4AAP) 0.075 mol·ℓ-1 (Sigma) and dopamine (DP) 0.15 mol·ℓ-1 (Sigma) aqueous solutions were prepared daily. The solutions were protected from light after the preparation and during their utilisation in the analytical experiments.
A solution of sulphuric acid 0.25 mol·ℓ-1 was used as carrier in the flow system. A 100 mg·ℓ-1 stock solution of V(V) was prepared by dissolving the appropriate amount of ammonium vanadate in hydrochloric acid 0.01 mol·ℓ-1. Standard solutions of V(V) were prepared daily from the stock solution by suitable dilutions in water.
The methodology was applied to ten water samples and to a waste water reference certified sample (LGC Promochem, SPS-WW2; 0.500±0.005 mg·ℓ-1). A solution of disodium ethylenediamine tetraacetic acid (EDTA) 3.5x10-5 mol·ℓ-1 was used as masking agent in the analysis of the reference sample.
Apparatus
Spectrophotometric measurements were made in a 6300 Jenway spectrophotometer set at 565 nm and equipped with a 18 µℓ flow cell (Helma 178.711QS, Müllheim, Balden, Germany). Analytical signals were recorded on a Kipp & Zonen BD 111 strip chart recorder or by a computer equipped with a convenient interface.
The SIA system (Fig. 1) consisted of a Gilson Minipuls 3 peristaltic pump equipped with a PVC pumping tube (1.2 mm i.d.) and an 8-port multi-position Vici Valco selection valve. Manifold components were connected by means of PTFE tubing, 0.8 mm i.d., which was also used for the holding and reaction coil (4 and 1.75 m, respectively).
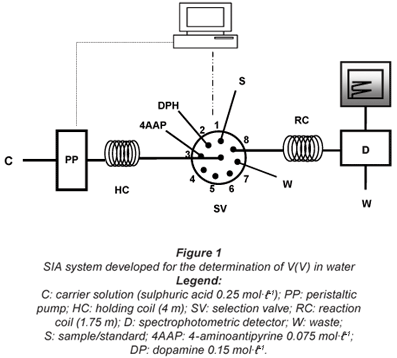
Analytical system control, including operation of the peristaltic pump and selection valve was achieved by means of an Advantech PCL 711B interface card and a Pentium-I based microcomputer. Software was developed in Microsoft Quick-Basic and permitted control of flow rate, flow direction, valve position, sample and reagent volume as well as data acquisition and processing.
Sequential injection procedure
To prepare the SIA system for the analytical determinations a small preliminary procedure was performed in order to fill the connecting tubing with carrier solution and the inlets of the selection valve with the respective solutions. This procedure involved the propulsion of carrier solution through Port 8 of the selection valve to fill the tubing and the flow cell and establish a baseline in the detector. Then a fixed amount of each solution was aspirated by the respective inlet to the holding coil and then by flow reversal the solution was sent to waste through Port 7.
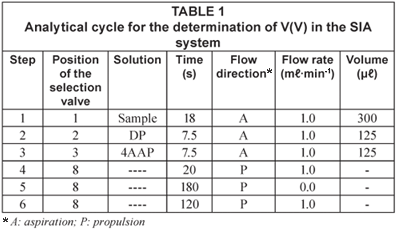
The analytical cycle (Table 1) began with the sequential aspiration of 300 µℓ of sample (Step 1), 125 µℓ of DP (Step 2) and 125 µℓ of 4AAP (Step 3) to the holding coil. The reaction zone, thus obtained, was propelled in the direction of the spectrophotometric detector (Step 4) and stopped in the reaction coil during 180 s (Step 5) in order to increase the sensitivity of the determination. After the stop period the reaction zone was sent to the detector (Step 6) and a signal, of absorbance proportional to the concentration of V(V), was registered.
Results and discussion
All studies related to the optimisation of the physical and chemical parameters involved in the determination of V(V), in the SIA automatic system, as well as the results from V(V) determination in water and their validation, are described below.
Optimisation of the physical and chemical parameters
Regarding the optimisation of the analytical procedure, several studies were performed with the aim of investigating the influence of sample and reagents volume, reagents concentration, order of aspiration, carrier flow rate, reaction coil length and configuration and carrier solution on the formation of the coloured product and consequently on the respective absorbance signal.
The SIA methodology developed for the determination of V(V) in water was based on a reaction presented by Kumar et al. (2007) in which the reagent MBTH was changed by 4AAP. The reaction involved the oxidation of DP by V(V) in acid medium and the coupling of the formed ceto-intermediary with 4AAP to form a violet product that was monitored at 565 nm.
The pH of the reaction media is of the most importance in this reaction since it determines the vanadium species in solution, influencing directly the sensitivity of the determination. In a SIA system, after the aspiration of sample and reagent zones mutual dispersion of these zones with the carrier is achieved by flow reversal, so that the carrier solution determines strongly the conditions of the reaction. Thus, solutions of sulphuric, nitric and hydrochloric acids with concentrations between 0.05 and 0.35 mol·ℓ-1 were tested as carrier solutions in the automatic system. The studies revealed that sulphuric acid 0.25 mol·ℓ-1 provided the best conditions for the reaction leading to an enhancement of about 42%, in terms of sensitivity, when compared to the other tested acids, in the same conditions. It was found that the analytical signals increased with the concentration of sulphuric acid up to 0.25 mol·ℓ-1 and that higher concentrations did not lead to a significant increase in sensitivity, so that the studies proceeded with that concentration.
The influence of sample volume was studied between 150 and 350 µℓ and it was observed that with the increase of volume up to 200 µℓ there was a sensitivity increase of about 3 times. Even though it was verified that the analytical signals increased with the volume of sample up to 350 µℓ, volumes higher than 300 µℓ led to irregular peaks revealing mixing problems. Thus, further studies were performed with 300 µℓ of sample.
The assessment of the influence of DP volume and concentration was carried out due to their importance to the oxidation reaction. Volumes of DP between 75 and 150 µℓ were tested and even though the analytical signal increased with the volume up to 150, this volume was shown to be excessive resulting in a poor mixture of the aspirated zones so that the studies proceeded with 125 µℓ of DP.
Regarding the concentration of DP, solutions with concentrations in the range of 0.05 to 0.20 mol·ℓ-1 were studied. The sensitivity of the determination increased about 50% with increased concentration in the considered concentration range. However, when the assays were performed with DP 0.20 mol·ℓ-1, there was a loss of linearity for V(V) concentrations above 3.0 mg ℓ-1, so that the remaining studies were conducted with DP 0.15 mol·ℓ-1.
The volume and concentration of 4AAP were studied in order to establish the best conditions for the coupling reaction. The effect of 4AAP volume was investigated between 75 and 200 µℓ. The determination was performed with 125 µℓ of 4AAP since higher volumes lead to an irregular distribution of the colour product in the reaction zone and peak irregularities.
Solutions of 4AAP with concentration between 0.05 and 0.15 mol·ℓ-1 were tested. The increase of the solution viscosity related to the increase of concentration hindered the mixture of the solutions online, so that for concentrations above 0.075 mol·ℓ-1 there was the formation of double peaks. Regarding sensitivity of the determination the best results were accomplished with 4AAP 0.075 mol·ℓ-1 so that further studies were conducted with a 4AAP solution with that concentration.
With the view of augmenting the sensitivity, a complementary test that consisted of aspirating a reagent solution obtained by the mixture of DP and 4AAP (in the optimised concentrations) was performed. Even though the sensitivity of the reaction increased slightly, the solution consisting of the two reagents was not stable through the working day, so that the studies proceeded with the aspiration of solutions of each individual reagent.
An important feature affecting system performance was the sample/reagent mixing and reaction zone homogenisation (Marshall and van Staden, 1992). In a SIA system these aspects are, to a great extent, determined by the sequence of aspiration of sample and reagents into the holding coil, which dictates how the different zones will mutually inter-disperse or which zone will endure a higher degree of dispersion. The best results in terms of mixing efficiency and signal magnitude were obtained when the sample was first introduced in the holding coil followed by DP and then 4AAP. This sequence allows an efficient mixing of the solutions and respects the natural sequence of the reaction in which DP is oxidised by V(V) and the formed intermediary reacts with 4AAP.
In a SIA system, due to the bidirectional nature of the flow, it is possible to implement stop periods with the aim of increasing the reaction time and consequently the sensitivity of the determinations (Ruzicka and Gubeli, 1991). Thus, after the aspiration of sample and reagents to the holding coil, the reaction zone was sent to the reaction coil and a stop period was introduced in the analytical cycle. The effect of stop periods of 30 to 240 s was tested and the sensitivity of the determination increased about 2.5 times with the stop period until 180 s. Longer periods did not lead to significant change of the analytical signals so that, as a compromise between sensitivity and sampling rate, a stop period of 180 s was introduced in the analytical cycle.
The better residence time of the reaction zone on the system was further evaluated by determining the effect of the reaction coil length and configuration on the analytical signals. The analytical signal increased with the reactor length until 1.75 m, showing that this coil led to the best conditions to implement the stop period without increasing significantly the dilution of the reaction product. Of the different tested configurations (straight, coiled, figure 8), figure 8 reactors led to higher analytical signals in the studied concentration range, confirming the fact that they originate lower dispersion of the reaction zone on its way to the detector. The influence of propulsion flow rate was evaluated between 0.8 and 2 mℓ·min-1 and the obtained results showed that the analytical signals increased with the flow rate up to 1 ml·min-1, showing that the residence time was excessive for lower flow rates. The determination was performed propelling the reaction zone to the detector at 1 mℓ·min-1.
Figures of merit
After the optimisation of all the parameters affecting the analytical signal, the developed methodology was evaluated with V(V) concentrations between 0.5 to 5.0 mg·ℓ-1 and linear calibration plots were obtained. The typical calibration plot was Abs (UA) = 0.0902 (±0.0029) C (mg·ℓ-1) + 0.06 (± 0.0066), R2=0,994 (Abs: absorbance; C: V(V) concentration). The detection limit of the determination, calculated as 3σ of the blank signal (Miller, 1991), was 0.39 mg·ℓ-1. The optimised procedure allowed performing about 10 determinations·h-1.
Interferences
Considering that the developed methodology was to be applied in the analysis of wastewater and polluted environmental samples it was important to assess the potential interfering effect of several species usually present in this kind of sample.
Standard solutions with a fixed amount of V(V) (1 mg·ℓ-1) and increasing concentrations of the potential interfering species were analysed by the automatic SIA methodology. A species was considered as non-interfering when the analytical signal variation relative to the one obtained in its absence was lower than 3%.
The results showed that Fe(III), Cr(VI), Cu(II), Ce(VI) and NO2- exhibit tolerance limits of 0.10 mg·ℓ-1 and that Pb2+, Na+, SO42-, NO3- and PO43- only affect the determination when present in concentrations above 100 mg·ℓ-1.
Analysis of water samples
Ten water samples and a certified reference wastewater sample were analysed for V(V) content immediately after sampling, by the proposed SIA methodology. The results showed that the V(V) content of tap water samples was below the detection limit of the methodology and so they were spiked to mimic contaminated samples.
The validation of the developed automatic procedure was performed through recovery assays in which different amounts of V(V) were added to the tap water samples and through the analysis of a reference wastewater sample. The recovery values (Table 2) ranged between 98.1 and 107.6%, indicating that there are no matrix effects. On the other hand, the result of the analysis of the wastewater reference sample was in good agreement with the certified value (0.50 mg·ℓ-1), with a relative deviation of 2.3%.
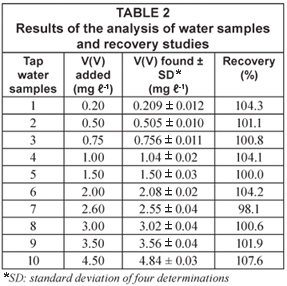
The precision of the methodology was evaluated in terms of relative standard deviation (rsd%) of fifteen determinations of two samples with V(V) concentration of 0.50 mg·ℓ-1 and 3.0·mg ℓ-1. The obtained rsd values (1.56 and 1.44% respectively) demonstrate the excellent repeatability of the automatic methodology.
Conclusions
The developed SIA system for the determination of V(V) in environmental polluted waters was shown to be simple, versatile and easy to operate, due to its peculiar mode of operation based on flow reversal. It allowed the analysis of a significant number of samples without system recalibration and little intervention from the operator. The introduction of a stop period in the analytical cycle for the determination of V(V) automatic procedure confirmed the versatility of the selection valve as central device of the system.
Relative to the existing procedures for the determination of V(V) in environmental waters, the developed methodology, even though it does not present evident advantages in terms of sensitivity, was shown to be faster and more economical, allowing the fast determination of V(V) with very simple equipment, reduced consumption of reagents and minimal production of effluent. The analysis involved the consumption of 1.9 mg of 4AAP and 2.9 mg of DP and the production of 2.35 mℓ of effluent per determination. The reduction of reagent consumption and effluent production, associated with the computer control of all the operations, represents the main advantages of the proposed procedure relative to the existing FIA methodologies for the determination of V(V) in environmental waters. Furthermore, the simplicity of the developed SIA system is an important advantage in comparison with some of the referred FIA procedures whose applicability is limited by the high cost of the involved instruments (Wuilloud et al., 2001; Yang et al., 1996).
The successful analysis of a wastewater reference sample confirmed the applicability of the proposed system to wastewater samples.
The features presented above, in association with the portable nature of the developed SIA system for the determination of V(V), result in an interesting alternative to other available proposals, with the developed SIA system being a good option for routine analysis of wastewaters and highly polluted environmental waters due to the possibility of performing in situ determinations.
References
ABBASPOUR A and MIRZAJANI R (2006) Application of β-correction method and partial least squares for simultaneous determination of V(IV) and V(V) in surfactant media. Spectrochim. Acta A 64 646-652. [ Links ]
CHEN ZL and OWENS G (2008) Trends in speciation analysis of vanadium in environmental samples and biological fluids – a review. Anal. Chim. Acta 607 1-14. [ Links ]
DEB MK, PATEL KS and MISHRA RK (1991) Extraction of vanadium(V) with hydroxyamidine in the presence of adductants and its spectrophotometric determination with diphenylcarbazide. Int. J. Environ. An. Ch. 43 209-217. [ Links ]
ENSAFI AA and KAZEMZADEH A (1996) Determination of vanadium by its catalytic effect on the oxidation of gallocyanine with spectrophotometric flow injection analysis. Microchem. J. 53 139-146. [ Links ]
KAMAVISDAR A and PATEL KS (2000) Extraction spectrophotometric determination of vanadium in industrial waste water. J. Chin. Chem. Soc. 47 1161-1164. [ Links ]
KLAASSEN CD (2008) Casarett & Doull's Toxicology: The Basic Science of Poison (7th edn.). Mc.Graw Hill Medical. 971-972. [ Links ]
KUMAR KS, KANG SH, SUVARDHAN K and KIRAN K (2007) Facile and sensitive spectrophotometric determination of vanadium in various samples. Environ. Toxicol. Pharmacol. 24 (1) 37-43. [ Links ]
LENEHAN CE, BARNETT NW and LEWIS SW (2002) Sequential injection analysis. Analyst 127 997-1020. [ Links ]
LI JJ, DU JX and LU JR (2002) Flow injection electrogenerated chemiluminescence determination of vanadium and its application to environmental water sample. Talanta 57 53-57. [ Links ]
MARSHALL GD and VAN STADEN JF (1992) Operational parameters affecting zone penetration in sequential injection analysis. Proc. Cont. Qual. 3 251-261. [ Links ]
MATSUOKA S, YOSHIMURA K and TATEDA A (1995) Application of ion-exchanger phase visible light absorption to flow analysis. Determination of vanadium in natural water and rock. Anal. Chim. Acta 317 207-213. [ Links ]
MILLER JN (1991) Basic statistical methods for analytical chemistry. Part 2. Calibration and regression methods. Analyst 16 3-14. [ Links ]
MOHAMED AA and FAWY KF (2000) Catalytic determination of vanadium based on the bromate oxidative-coupling reaction of metal with phloroglucinol. Mikrochim. Acta 134 229-235. [ Links ]
MOYANO S, POLLA G, SMICHOWSKI P, GÁSQUEZ JA and MARTINEZ LD (2006) On-line pre-concentration and determination of vanadium in tap and river water samples by flow injection-inductively coupled plasma-optical emission spectrometry (FI-ICP-OES). J. Anal. At. Spectrom. 21 422-426. [ Links ]
MUKHERJEE B, PATRA B, MAHAPATRA S, BANERJEE P, TIWARI A and CHATTERJEE M (2004) Vanadium – an element of atypical biological significance. Toxic. Lett. 15 135-143. [ Links ]
NAKANO S, TAGO M and KAWASHI T (1989) Catalytic determination of nanogram amounts natural water by flow injection analysis. Anal. Sci. 5 69-72. [ Links ]
NAKANO S, SAKAMOTO K, TAKENOBU A and KAWASHIMA T (2002) Flow-injection chemiluminescent determination of vanadium(IV) and total vanadium by means of catalysis on the periodate oxidation of purpurogallin. Talanta 58 1263-1270. [ Links ]
PYRZYŃSKA K and WIERZBICKI T (2004) Determination of vanadium species in environmental samples. Talanta 64 823-829. [ Links ]
PYRZYŃSKA K (2005) Recent developments in spectrophotometric methods for determination of vanadium. Mikrochim. Acta 149 159-164. [ Links ]
RUZICKA J and MARSHALL GD (1990) Sequential injection: a new concept for chemical sensors, process analysis and laboratory assays. Anal. Chim. Acta 237 329-343. [ Links ]
RUZICKA J and GUBELI T (1991) Principles of stopped-flow sequential injection analysis and its application to the kinetic determination of traces of a proteolytic enzyme. Anal. Chem. 63 1680-1685. [ Links ]
SHIOBARA T, TESHIMA N, KURIHARA M, NAKANO S and KAWASHIMA T (1999) Catalytic flow injection determination of vanadium by oxidation of N-(3-sulfopropyl)-3,3',5,5'- tetramethylbenzidine using bromate. Talanta 49 1803-1809. [ Links ]
TAYLOR MJC and VAN STADEN JF (1994) Spectrophotometric determination of vanadiurn(IV) and vanadium(V) in each other's presence – Review. Analyst 119 1263-1276. [ Links ]
WUILLOUD RG, WUILLOUD JC, OLSINA RA and MARTINEZ LD (2001) Speciation and pre-concentration of vanadium(V) and vanadium(IV) in water samples by flow injection-inductively coupled plasma optical emission spectrometry and ultrasonic nebulisation. Analyst 126 715-719. [ Links ]
YAMANE T, OSADA Y and SUZUKI M (1998) Continuous flow system for the determination of trace vanadium in natural waters utilizing in-line pre-concentration: separation coupled with catalytic photometric detection. Talanta 45 583-589. [ Links ]
YANG KL, JIANG SJ and HWANG TJ (1996) Determination of titanium and vanadium in water samples by inductively coupled plasma mass spectrometry with on-line pre-concentration. J. Anal. At. Spectrom. 11 139-143. [ Links ]
YEH CF and JIANG SJ (2004) Speciation of V, Cr and Fe by capillary electrophoresis-bandpass reaction cell inductively coupled plasma mass spectrometry. J. Chromatogr. A 1029 255-261. [ Links ]
EMBERYOVÁ M, JANKOVIČ R, HAGAROVÁ I and KUSS H-M (2007) Electrothermal atomic absorption spectrometric determination of vanadium in extracts of soil and sewage sludge certified reference materials after fractioning by means of the Communities Bureau of Reference modified sequential extraction procedure. Spectrochim. Acta B 62 509-513. [ Links ]
Received 24 June 2008; accepted in revised form 15 January 2009.
* To whom all correspondence should be addressed.
+351 222078939; fax: +351 222078961;
e-mail: ppinto@ff.up.pt

