










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Orthopaedic Journal
versão On-line ISSN 2309-8309
versão impressa ISSN 1681-150X
SA orthop. j. vol.20 no.3 Centurion 2021
http://dx.doi.org/10.17159/2309-8309/2021/v20n3a8
ORTHOPAEDIC ONCOLOGY AND INFECTIONS
Malignant transformation in an 11-year-old child with multiple hereditary exostosis
Janet L de StadlerI,*; Neil KrugerII; Shivani SinghI; Ebrahim BanderkerIII; Stewart Dix-PeekIV; Komala PillayI
IDivision of Anatomical Pathology, Groote Schuur and Red Cross War Memorial Children's Hospital, National Health Laboratory Services, University of Cape Town, Cape Town, South Africa
IIDepartment of Orthopaedics, Groote Schuur and Red Cross War Memorial Children's Hospital, Life Kingsbury Hospital, University of Cape Town, Cape Town, South Africa
IIIDepartment of Diagnostic Radiology, Red Cross War Memorial Children's Hospital, University of Cape Town, Cape Town, South Africa
IVPaediatric Orthopaedic Unit, Red Cross War Memorial Children's and Maitland Cottage Hospitals, University of Cape Town, Cape Town, South Africa
ABSTRACT
BACKGROUND: Multiple hereditary exostosis (MHE) is a rare autosomal dominant disorder predisposing to the development of multiple osteochondromas. Malignant transformation is an uncommon complication of osteochondromas and is especially rare in the paediatric population. Making a diagnosis of malignant transformation is recognised as a challenge globally
METHODS: We obtained informed consent and ethics approval prior to reviewing the hospital file, radiology and pathology of our index patient, as well as conducting a directed literature search
RESULTS: An 11-year-old male with MHE presented with new onset pain in the right leg with an associated inability to weight bear. Plain radiographs and magnetic resonance imaging (MRI) showed features consistent with malignant transformation. The child underwent a Malawer 1 resection of the proximal fibula with no complications. The pathology confirmed a grade 1 secondary peripheral chondrosarcoma (CS) arising in an osteochondroma
The rate of malignant transformation in MHE is as high as 36.3% in select specialist tertiary centres. Ninety per cent of the resultant malignancies are chondrosarcomas. Malignant transformation before the age of 20 years is exceptional. Plain radiology is routinely used for monitoring of patients with MHE. Other modalities exist to assess for cartilage cap thickness, a much-debated criterion of malignant change. Pathology is essential for confirmation of malignant transformation as well as to exclude high grade lesions. Treatment is wide local excision (WLE) with limb-sparing surgery and long-term follow-up to detect for local recurrences.
CONCLUSION: The malignant transformation of osteochondromas occurs more frequently in individuals with MHE and may even arise in the paediatric population. In the presence of suspicious clinical or radiological features, en-bloc surgical resection and histopathological correlation is mandatory to make the diagnosis. We encourage a multidisciplinary team approach with collaboration between the orthopaedic surgeon, radiologist and pathologist
Level of evidence: Level 5
Keywords: multiple hereditary exostosis (MHE), chondrosarcoma, osteochondroma, malignant transformation
Introduction
Multiple hereditary exostosis (MHE), also known as multiple osteochondromas (MO), is a rare autosomal dominant disorder with a prevalence of approximately 1 in 50 000 in the general population.1 The majority have germline mutations in either the EXT1 or EXT2 tumour suppressor genes, which encode proteins involved in chondrocyte proliferation and differentiation.2,3
Osteochondromas are variably sized, benign cartilaginous neoplasms arising from the external, juxtaepiphyseal region of bones which have formed by endochondral ossification. They may be sessile or pedunculated and are composed of an external cartilage cap, underlying cortical bone, and an innermost medullary cavity which merges with that of the bone of origin.1,4 The most frequently affected sites include the distal femur, proximal tibia and humerus.1
MHE is more likely to affect males and is characterised by multiple osteochondromas, often accompanied by short stature with or without angular or limb length deformities. Individuals with a family history and at least two juxtaepiphyseal long bone osteochondromas are diagnosed clinically. Genetic testing is not required. These osteochondromas may present soon after birth and continue to grow throughout childhood until the growth plates close. The majority of affected individuals are diagnosed by the age of 12 years.1,2
Osteochondromas are frequently painless and slow growing, mostly causing a cosmetic deformity.4 However, the direst complication is that of malignant transformation, invariably due to a secondary chondrosarcoma (CS).4 This transformation, as discussed later, is especially rare in children.2
Case report
An 11-year-old male, known to the Orthopaedic Department at Red Cross War Memorial Children's Hospital with MHE, presented with new onset right knee pain and an associated inability to fully weight bear for three days. There were no associated fevers or systemic upset. He was first diagnosed at the age of 4 years. By 10 years he had osteochondromas in both proximal humeri, both femurs (proximally and distally), both proximal tibias, the right distal tibia, the right mid-distal ulna and the right proximal fibula.
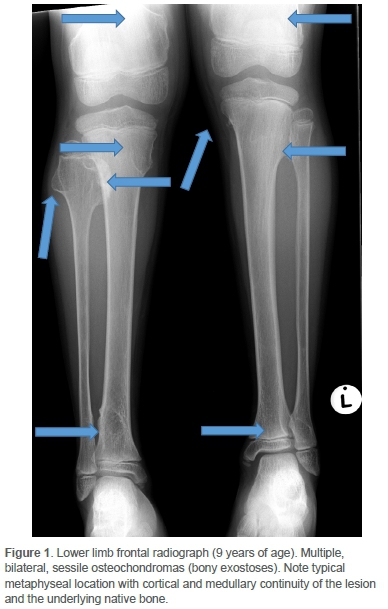
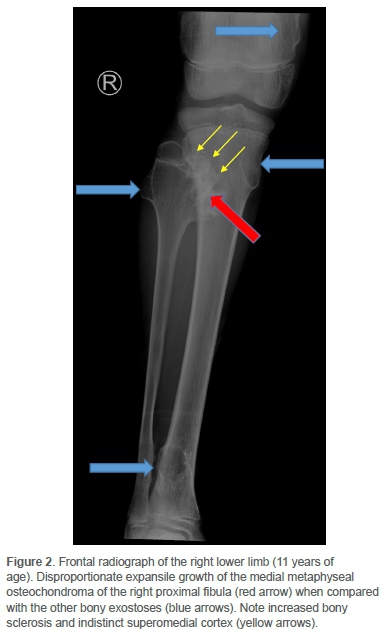
Clinically he had an antalgic gait and the pain was localised to the distal medial femoral condyle. There was no effusion. He achieved full extension, but flexion beyond 90° was resisted. Concern regarding sarcomatous change was raised on plain radiographs of the knee. Comparison with images two years prior yielded sinister interval morphological change and exuberant growth of a singular osteochondroma located at the medial metaphysis of the right proximal fibula (Figures 1 and 2).
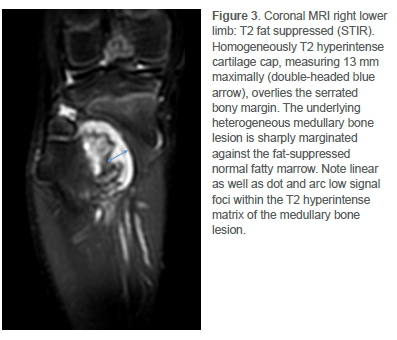
Pre- and post-contrast enhanced magnetic resonance imaging (MRI) then demonstrated aggressive bone changes commensurate with malignant transformation (Figures 3 and 4). Most notable was the irregularity of the overlying cartilage cap and abrupt margination of an enhancing T2 hyperintense underlying intramedullary soft tissue mass.
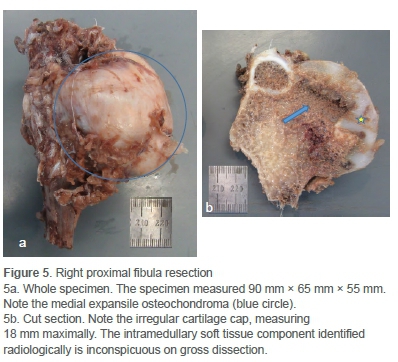
The child underwent a Malawer 1 resection, an en bloc yet marginal resection of the proximal fibula and tumour, sparing the common peroneal nerve (CPN) and anterior tibial artery.5 The resected specimen was large (Figure 5a), and the CPN had to be mobilised near the full length of the incision to ensure complete excision. The bulbous growth of the tumour both into and below the proximal tibiofibular joint notched into the adjacent tibia and made deep dissection difficult. The proximal tibiofibular joint and a small segment of adjacent tibial metaphysis were excised. The lateral collateral ligament complex, having been initially detached from the proximal fibula, was whipstitched with a 3 ethibond and reattached to the proximal lateral tibia via transosseous drill holes and periosteal suturing. The tourniquet was released to ensure meticulous haemostasis. The wound was closed in layers and a pressure dressing applied. He was discharged home in an above-knee backslab after an uneventful hospital stay.
He attended outpatient physiotherapy and was reviewed at two, four, six and ten weeks post-surgery. Wound healing was slow, with some sloughing and necrosis of the edges. By ten weeks, he was partially weight-bearing without assistance and had 10-120° range of movement at the knee. He was completely well at his five-month follow-up, and was asked to return in six months to continue annual surveillance.
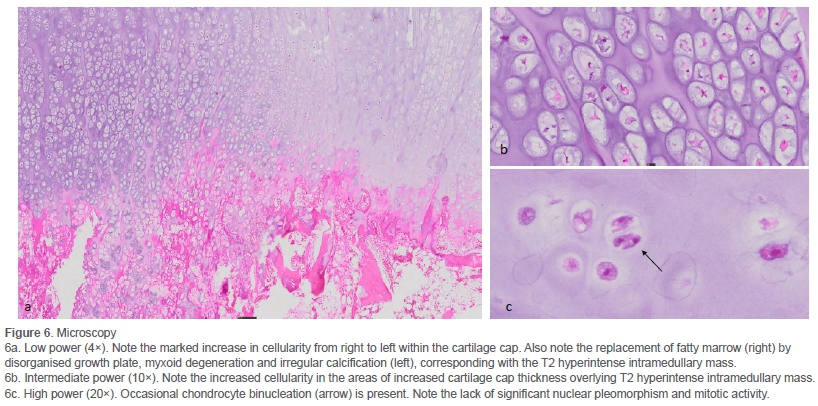
Cut section of the specimen confirmed the irregularity of the overlying cartilage cap (Figure 5b). Microscopy confirmed a grade 1 secondary peripheral CS/atypical cartilaginous tumour (ACT) arising in an osteochondroma (Figure 6).
Discussion
The rate of malignant transformation in MHE is between 3% and 5%,4 although figures are as high as 36.3% in specialist tertiary centres which see a preselected high-risk population.6 The rate is higher for centrally located lesions, for example, in the pelvis, compared to peripherally located lesions, such as around the knee.7-9 This may be the result of early excision of peripheral lesions due to more frequent benign complications.9
The average time to malignant transformation is 9.8 years from initial diagnosis. Ninety per cent of the resultant malignancies are CS.8 ACT is the preferred term for a grade 1 peripheral CS arising in the appendicular skeleton and, apart from their location, are identical to the axial counterpart grade 1 peripheral CS.1 Making a diagnosis of a secondary CS is recognised as a challenge globally,6 and emphasis is placed on a multidisciplinary team approach with collaboration between the orthopaedic surgeon, radiologist and pathologist.7
Clinical
Malignant transformation before the age of 20 years is exceptional4 and most data are from case reports or series.3,6,9 One may suspect malignant transformation in an adult presenting with an enlarging osteochondroma and/or pain,2,6 or due to changes noted at annual review.6 In contrast, growth and pain of a pre-existing lesion in the skeletally immature population are not as concerning.3 Case reports, however, do describe mild pain, gait abnormalities, and a clinically enlarging lesion as features prompting investigation and eventual diagnosis of secondary CS in children.3 In this case, the non-localising pain was likely a red herring prompting imaging which then raised the suspicion for further investigation. This was three months ahead of his scheduled annual review. As the excised lesion was a grade 1 ACT, later detection at this date would have been unlikely to have affected the outcome.
Radiology
Plain radiology is routinely used for monitoring of patients with MHE for malignant change. Features to note include irregularity of the surface, foci of radiolucency, heterogeneity, non-uniform calcification, erosion of the adjacent bone and an associated soft tissue mass.4,6 MRI is necessary to determine the cartilage cap thickness in suspected cases. In addition, MRI can more accurately delineate a soft tissue mass, and allow for surgical planning.6,8 Ultrasound scanning may also be used to measure cartilage cap thickness.4
While a cartilage cap thickness of 2 cm or greater has been suggested to be 100% sensitive and 98% specific for secondary CS using MRI,8 a thickness of 1.5 cm or greater is still considered sufficiently concerning.3,4 Furthermore, several conflicting case reports of CS show measurements between 0.5 and 1.5 cm.6,7 Some authors propose that the cartilage cap quality may be more important than the thickness,6 while others advocate that cartilage cap thickness should not be used as an indicator of malignant transformation in the paediatric population at all.4 In this case, placing too much reliance on a cartilage cap cut-off of 2 cm would have resulted in misdiagnosis and delayed treatment.
Pathology
As in this case, up to 85% of secondary CS are grade 1 lesions.4,6,7,9 Distinguishing a grade 1 peripheral CS/ACT from an osteochondroma on histology is subjective. Features such as nodularity, binucleate chondrocytes, myxoid degeneration and irregular calcifications may frequently be present in both lesions.7 An infiltrative growth pattern6 and invasion of surrounding soft tissue and bone cortex, when present, may assist in the diagnosis.4 In our case, the clear transition from low to high cellularity within the thickened cartilage cap, as well as the infiltration of fatty marrow by myxomatous cartilage and irregular calcifications in the areas corresponding with the T2 hyperintensity on MRI were compatible with transformation.
Microscopy is also essential to exclude a grade 2, 3 or dedif-ferentiated CS by observing the lack of mitotic activity, nuclear pleomorphism and a malignant spindle cell component.7 In addition, it is useful for subtyping, the majority of which are CS, not otherwise specified (NOS). Alternate subtypes may impact both treatment and prognosis.10
Management and prognosis
CS ideally require wide local excision (WLE) with limb-sparing surgery to prevent recurrence. Care must be taken to excise the entire perichondrium and prevent myxomatous cartilage leak into the surgical bed to minimise the risk of recurrence.3,4,6,7 Marginal excision may be considered in surgically challenging locations.6,7 Both chemotherapy and radiotherapy are of little benefit as CS are mostly resistant.4,6,10
If there is uncertainty of malignant transformation, close follow-up with serial imaging and timely surgical treatment is appropriate.2,3,8 Pre-excision biopsy is not advised, unless a high-grade malignancy is suspected.3
Long-term follow-up is essential to detect local recurrences, which often occur in the first Ave years after surgery and occur slightly more often in individuals with MHE.6 Recurrence rates in WLEs are low, between 0% and 15%, whereas rates in marginal or intralesional resections are much higher, between 57% and 78%.4 Regardless, annual clinical review and radiological screening, ideally MRI, is recommended for all individuals with MHE.1,2 The majority of deaths are due to complications of a local recurrence, highlighting the need for adequate surgical excision. Importantly, repeated excisions for recurrent lesions can result in eventual progression to a higher-grade CS.6 The frequency of follow-up of cases with prior malignant transformation is not addressed by the literature reviewed.
Conclusion
The malignant transformation of osteochondromas occurs more frequently in individuals with MHE and may even arise in the paediatric population. The diagnosis is especially challenging in this age group. In the presence of suspicious clinical or radiological features, en-bloc surgical resection and histopathological correlation is mandatory to make the diagnosis. Long-term follow-up is essential to detect recurrences. We encourage a multidisciplinary team approach with collaboration between the orthopaedic surgeon, radiologist and pathologist.
Acknowledgement
We thank Jurgen Geitner, Senior Technical Officer at the University of Cape Town (UCT) Pathology Learning Centre, for providing the whole slide images for the micrographs.
Ethics statement
Prior to commencement of the study, ethical approval was obtained from the Human Research Ethics Committee, Faculty of Health Sciences, University of Cape Town. HREC REF: 675/2020, and written informed consent was obtained from the legal guardian.
Declaration
The authors declare authorship of this article and that they have followed sound scientific research practice. This research is original and does not transgress plagiarism policies.
Author contributions
De Stadler JL: Primary author, conceptualisation, design, data collection, analysis, and manuscript preparation, critical revision for important intellectual content and final approval of the version submitted to the journal
Kruger N: Data collection, analysis, manuscript preparation, critical revision for important intellectual content and final approval of the version submitted to the journal Singh S: Data collection, critical revision for important intellectual content and final approval of the version submitted to the journal
Banderker E: Manuscript preparation, critical revision for important intellectual content and final approval of the version submitted to the journal
Dix-Peek S: Critical revision for important intellectual content and final approval of the version submitted to the journal
Pillay K: Critical revision for important intellectual content and final approval of the version submitted to the journal
ORCID
De Stadler JL https://orcid.org/0000-0001-6246-1716
Kruger N https://orcid.org/0000-0002-0929-2092
Dix-Peek S https://orcid.org/0000-0002-3382-8790
Pillay K https://orcid.org/0000-0003-1971-900X
References
1. Bovée JVMG, Hogendoorn PCW, Sangiorgi L. Genetic tumour syndromes of soft tissue and bone. In: Soft tissue and bone tumours [Internet]. 5th ed. Lyon: IARC; 2020. Available from: https://tumourclassification.iarc.who.int/chaptercontent/33/204. [ Links ]
2. Kivioja A, Ervasti H, Kinnunen J, et al. Chondrosarcoma in a family with multiple hereditary exostoses. J Bone Jt Surg - Ser B. 2000;82(2):261-66. Available from: https://europepmc.org/article/med/10755438. [ Links ]
3. Schmale GA, Hawkins DS, Rutledge J, Conrad EU. Malignant progression in two children with multiple osteochondromas. Sarcoma. 2010;2010:417105. Available from: https://europepmc.org/article/med/20467466 [ Links ]
4. Murphey MD, Choi JJ, Kransdorf MJ, et al. From the archives of the AFIP. Imaging of osteochondroma: Variants and complications with radiologic-pathologic correlation. Radiographics. 2000;20(5):1407-34. Available from: https://pubs.rsna.org/doi/full/10.1148/radiographics.20.5.g00se171407. [ Links ]
5. Malawer MM. Surgical management of aggressive and malignant tumors of the proximal fibula. Clin Orthop Relat Res [Internet]. 1984 Jun;(186):172-81. Available from: https://europepmc.org/abstract/MED/6723139. [ Links ]
6. Ahmed AR, Tan TS, Unni KK, et al. Secondary chondrosarcoma in osteochondroma: Report of 107 patients. Clin Orthop Relat Res. 2003;(411):193-206. Available from: https://pubmed.ncbi.nlm.nih.gov/12782876/. [ Links ]
7. De Andrea CE, Kroon HM, Wolterbeek R, et al. Interobserver reliability in the histopathological diagnosis of cartilaginous tumors in patients with multiple osteochondromas. Mod Pathol. 2012;25(9):1275-83. Available from: https://ora.ox.ac.uk/objects/uuid:72df7907-561e-46ff-9daa-e594680191b0. [ Links ]
8. Bernard SA, Murphey MD, Flemming DJ, Kransdorf MJ. Improved differentiation of benign osteochondromas from secondary chondrosarcomas with standardized measurement of cartilage cap at CT and MR imaging. Radiology. 2010;255(3):857-65. Available from: https://pubmed.ncbi.nlm.nih.gov/20392983/. [ Links ]
9. Altay M, Bayrakci K, Yildiz Y, et al. Secondary chondrosarcoma in cartilage bone tumors: Report of 32 patients. J Orthop Sci. 2007;12(5):415-23. Available from: https://pubmed.ncbi.nlm.nih.gov/17909925/. [ Links ]
10. Wu A-M, Li G, Zheng J-W, et al. Chondrosarcoma in a paediatric population: a study of 247 cases. J Child Orthop [Internet]. 2019 Feb;13(1):89-99. Available from: https://online.boneandjoint.org.uk/doi/10.1302/1863-2548.13.180109. [ Links ]
Received: August 2020
Accepted: November 2020
Published: August 2021
* Corresponding author: rbljan001@myuct.ac.za
Editor: Prof. Theo le Roux, University of Pretoria, Pretoria, South Africa
Funding: No funding was received for this study.
Conflict of interest: The authors declare they have no conflicts of interest that are directly or indirectly related to the research.


{kind=link}
{kind=link}