










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Orthopaedic Journal
versão On-line ISSN 2309-8309
versão impressa ISSN 1681-150X
SA orthop. j. vol.20 no.3 Centurion 2021
http://dx.doi.org/10.17159/2309-8309/2021/v20n3a7
ORTHOPAEDIC ONCOLOGY AND INFECTIONS
Phosphaturie mesenchymal tumour, 'non-phosphaturic' variant: a case report and review of the literature
Reubina WadeeI,*; Zwelithini LindaII; Abdullah IsmailI
IDepartment of Anatomical Pathology, University of the Witwatersrand/National Health Laboratory Service (NHLS), Johannesburg, South Africa
IIDepartment of Orthopaedic Surgery, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND: Phosphaturic mesenchymal tumours are uncommon neoplasms, usually associated with tumour-induced osteomalacia, that occur in middle-aged adults but have been reported at the extremes of age. The 'non-phosphaturic' variant is even rarer
METHODS: Herein, we describe the non-phosphaturic variant in a 12-year-old male who presented to the Department of Orthopaedic Surgery with a six-month history of painful progressive swelling of his right forearm in the absence of trauma. He had no other significant symptoms
RESULTS: The patient had normal serum calcium and phosphate levels on biochemical analysis. An inci-sional biopsy was performed and a histopathological diagnosis of a phosphaturic mesenchymal tumour, non-phosphaturic variant, was rendered. Six weeks later, he underwent surgical excision of a 15x15 cm soft tissue mass from his right forearm. He had an uneventful postoperative period and was discharged. He has been followed up at the outpatient department and has been free of tumour recurrence for over 18 months since his surgery with no evidence of osteomalacia and no other tumours
CONCLUSION: Phosphaturic mesenchymal tumours are rare mesenchymal neoplasms and their microscopically identical counterpart without phosphaturia, known as the 'non-phosphaturic' variant, is even more uncommon and may prove a greater diagnostic challenge. While the diagnosis may be confirmed by hypophosphataemia and phosphaturia secondary to the paraneoplastic phenomenon of tumour-induced osteomalacia, there may be instances, such as with our patient, where tumour-induced osteomalacia is absent. This case underscores the importance of clinicopathological correlation together with a wide differential diagnosis required to arrive at a correct diagnosis to facilitate appropriate patient management
Level of evidence: Level 5
Keywords: phosphaturic mesenchymal tumours, non-phosphaturic variant
Introduction
Phosphaturic mesenchymal tumours (PMTs) are rare neoplasms that are usually associated with renal phosphate loss and tumour-induced osteomalacia (TIO).1 Clinically, patients may have symptoms of hypophosphataemia and on thorough examination, small soft tissue or intraosseous masses in the extremities or the jaw may be identified.2,3 TIO occurs secondary to excessive production of tumour-associated fibroblast growth factor-23 (FGF23).4 This hormone is secreted by osteocytes and causes phosphaturia by inhibiting renal phosphate reabsorption and, by inhibiting activity of the 1a hydroxylase enzyme, results in decreased production of 1,25-hydroxyvitamin D.4,5 Occasionally, cases of PMT may not demonstrate TIO and as such, are referred to as 'non-phosphaturic' variants.5 A histological diagnosis in these circumstances may be confirmed by a comprehensive immunohistochemical panel excluding other tumours and by the detection of FGF23 transcripts using reverse transcription-polymerase chain reaction (RT-PCR) or in-situ hybridisation.5
Herein, we present a case of a 12-year-old patient who was diagnosed with this rare non-phosphaturic variant of PMT.
Case report
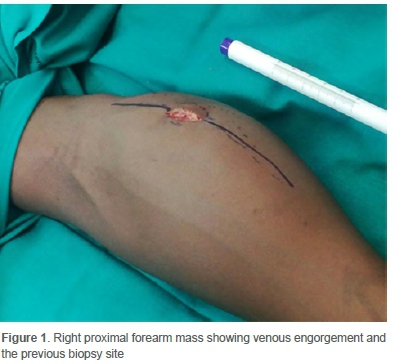
A 12-year-old male presented to the Department of Orthopaedic Surgery with a six-month history of painful progressive swelling of his right forearm. He reported no history of trauma to his forearm and had no other significant symptoms. On examination, he was clinically well and did not have any other masses. There was no neurological deficit in the right forearm. Local examination revealed a 15x15 cm mass on his right proximal forearm that was tender, non-pulsatile and immobile. Overlying skin changes in the form of prominent veins were noted (Figure 1).
Plain radiographs showed a large lesion with an osteoid matrix abutting the ulna (Figure 2). Magnetic resonance imaging (MRI) showed a large mixed cystic mass with foci of calcification and fluid-fluid levels. The mass covered the anteromedial aspect of the ulna and was in close association with the proximal ulna (Figure 3).


Relevant laboratory investigations showed the following:
• Calcium: 2.51 mmol/L (n=2.17-2.54 mmol/L)
• Magnesium: 0.87 mmol/L (n=0.66-0.91 mmol/L)
• Inorganic phosphate: 1.42 mmol/L (n=1.05-1.85 mmol/L)
The clinical differential diagnosis included juxtacortical osteochondroma, chondrosarcoma, synovial sarcoma, soft tissue sarcoma and traumatic myositis with cystic changes.
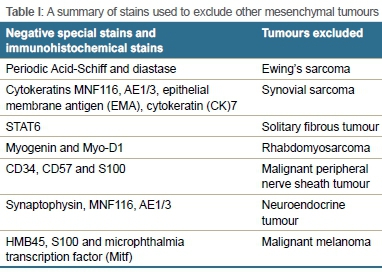
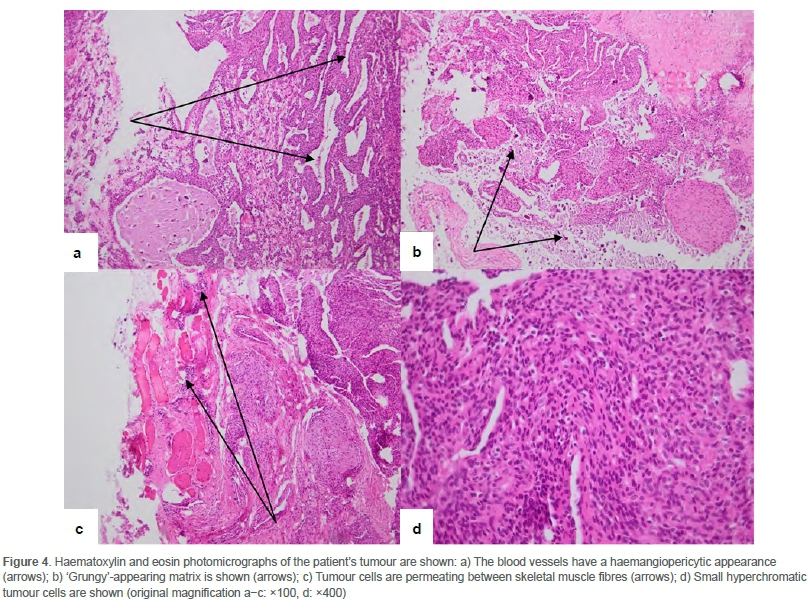
The patient initially underwent an incisional biopsy which was submitted for histopathological evaluation. Microscopically, the specimen consisted of fibrous connective tissue together with spicules of bone and a cellular, neoplastic infiltrate. The blood vessels had a haemangiopericytic appearance. The tumour was composed of small, hyperchromatic cells which had a spindled to stellate appearance with moderate amounts of eosinophilic cytoplasm. There was no mitotic activity or hypercellularity and areas of necrosis were absent. Foci of hyalinisation together with distinctive 'grungy'6,7 or flocculant-appearing calcified matrix, typical of PMTs, were seen. In areas, the tumour infiltrated surrounding skeletal muscle fibres (Figure 4). Both TIO-associated PMTs and non-phosphaturic variants show the same histological features.5 Table I summarises the special stains and immunohistochemical stains which were negative and thus excluded other mesenchymal tumours in the differential diagnosis. Focal, weak positive staining was noted on a CD99 stain. BCL2 was positive in tumour cells while scattered histiocytes were identified on a CD68 stain. These stains are not diagnostic of a particular tumour.
Tissue was submitted for assessment of the SYT-SSX1/2 transcripts which yielded a negative result, thus excluding a synovial sarcoma. In addition, fluorescence in-situ hybridisation (FISH) for 22q12 re-arrangement of the EWSR1 gene was performed and was negative, excluding Ewing's sarcoma. A diagnosis of a phosphaturic mesenchymal tumour, non-phosphaturic variant, was made.
Six weeks later, once a definitive diagnosis was made and theatre time was available, the patient underwent excision of the mass which was performed by a consultant orthopaedic surgeon. Intraoperatively, a fluid-filled mass was found (Figure 5). The tumour was firmly attached to the underlying muscles and focally to the periosteum of the ulna, but there was no bony erosion (Figure 5). During resection, the tumour ruptured, and white chalklike fluid extruded from the tumour. Macroscopically, the specimen consisted of a soft tissue mass which measured 80*60*30 mm. Numerous cystic areas and calcified regions were seen on the cut section of the tumour. Microscopic examination revealed identical features to the incisional biopsy. Viable tumour was identified at the peripheral margins of excision.

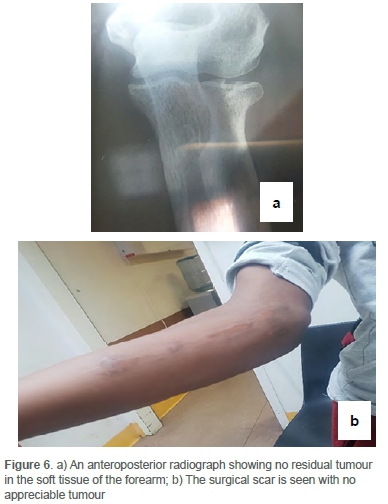
The patient had an uneventful postoperative course and was discharged. He has been followed-up on an outpatient basis for over 18 months and has no evidence of local recurrence on clinical examination (Figure 6). His follow-up included clinical examination together with serial chest X-rays and X-rays of his limbs at each follow-up visit, but he has not had additional MRI/CT scans or biochemical investigations.
Discussion
PMTs are uncommon tumours with fewer than 500 cases reported to date, most of which have been diagnosed in middle-aged adults.6 Tumours occurring at the extremes of age have, however, been documented.6 PMTs are neoplasms of uncertain histogenesis which may arise in bone or soft tissue.1,7 Soft tissue PMTs have a propensity to develop in the extremities while PMTs of bone tend to arise in the cranium, sinuses or appendicular skeleton.6 Patients often present with nonspecific symptoms and signs of TIO such as muscle pain, generalised weakness and pathological fractures due to excessive FGF23 production.7,8 FGF23 is a peptide that has an important role in phosphate reuptake in the kidneys via FGF receptor and a co-receptor (a-Klotho).6,9,10 Increased tumour-associated FGF23 results in phosphate wasting by decreased renal uptake of phosphate and increased renal excretion. Phosphate is required for several cellular metabolic functions such as energy production and intracellular signal transduction.10 As such, hypophosphataemia may result in nonspecific symptoms. The rare occurrence of PMT together with the insidious onset of symptoms often results in a delayed diagnosis of approximately three years.3,8
Microscopically, other mesenchymal neoplasms such as chondroblastoma, chondromyxoid fibroma, giant cell tumour of bone, solitary fibrous tumour, tenosynovial giant cell tumours, osteosarcomas and mesenchymal chondrosarcomas may be considered in the differential diagnosis of a PMT.4 In the present case, however, the other mesenchymal tumours noted above were excluded morphologically and immunohistochemically. Soft tissue Ewing's sarcoma and synovial sarcoma were excluded by immunohistochemical and molecular tests. The present non-phosphaturic variant of PMT did not show histological features of malignancy such as hypercellularity, nuclear pleomorphism, increased mitotic activity and necrosis, which have been described in current literature.6 PMTs do not have a specific immunohistochemical profile and as such, a broad panel of stains may be used to exclude other possible mesenchymal neoplasms, such as in the present case.
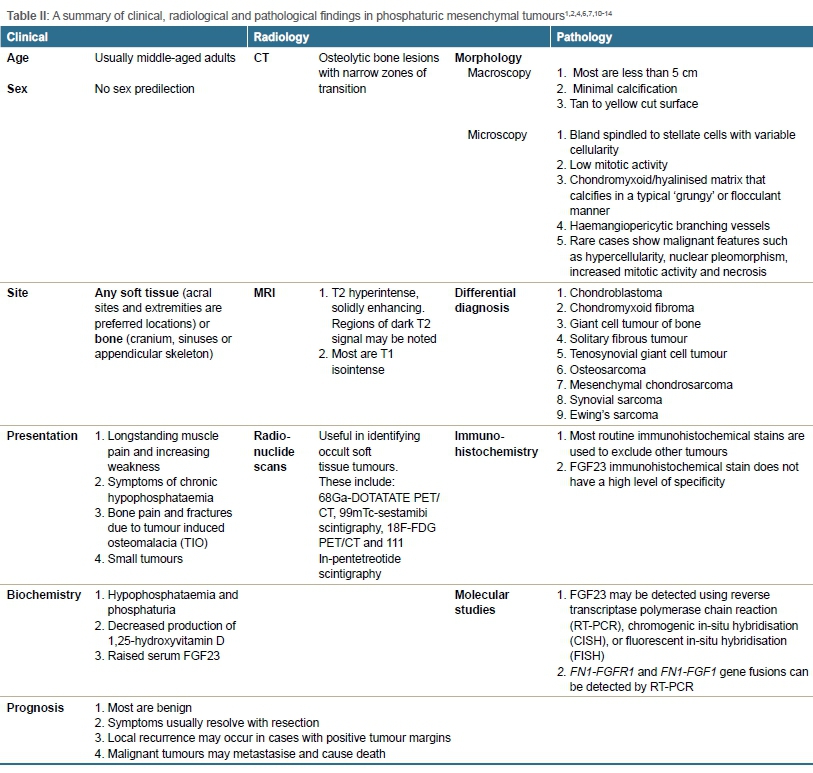
Biochemically, raised serum FGF23 levels together with hypo-phosphataemia may suggest a PMT if a patient has normal renal function. However, serum FGF23 levels may be compensated for or may not be sufficiently elevated for detection.11 This test is, however, not widely available globally. Gene fusions between fibronectin-1 (FN1) and fibroblast growth factor receptor-1 (FGFR1) as well as fusions between FN1 and fibroblast growth factor-1 (FGF1) have been identified by reverse transcriptase polymerase chain reaction (RT-PCR). While FGF23 may be detected in most PMTs, including non-phosphaturic variants, using RT-PCR, chromogenic in-situ hybridisation (CISH), or fluorescent in-situ hybridisation (FISH), thus confirming the histopathological diagnosis, these tests are often not available in most routine histopathology departments. It has been noted that FISH has demonstrated less sensitivity on archived material.1,12 While immunohistochemistry, however, is accessible to most pathology laboratories, the FGF23 immunohistochemical stain has not demonstrated a high level of specificity and is therefore not widely used. Thus, a multidisciplinary team approach including clinical, radiological and pathological findings are required to arrive at a definitive diagnosis.1Table II summarises clinical, radiological and pathological findings of PMTs.1,2,4,6,7,10-14
Wu et al. and Honda et al. have documented that for patients with TIO, complete excision of the tumour with negative margins is the preferred treatment.8,13 While the patient in the present case report did not have TIO, he did have a positive surgical excision margin which is known to increase the likelihood of local recurrence.7 He is, therefore, being followed up on an outpatient basis with clinical examination and serial chest and limb X-rays. It has been noted that patients who have tumours demonstrating uptake of octreotide on imaging studies may derive benefit from octreotide replacement therapy. Furthermore, early-stage clinical trials have suggested that antibodies to FGF23 may be advantageous for patients with TIO.8 Additional research is, however, required in this regard.
Conclusion
PMTs are rare mesenchymal neoplasms and their microscopically identical counterpart without phosphaturia, known as the 'non-phosphaturic' variant, is even more uncommon and may prove a greater diagnostic challenge. While the diagnosis may be confirmed by hypophosphataemia and phosphaturia secondary to the paraneoplastic phenomenon of TIO, there may be instances, such as with our patient, where TIO is absent. These tumours require thorough clinicopathological and radiological correlation together with a broad differential diagnosis to ensure accurate diagnosis and appropriate, timeous management of the patient.
Ethics statement
The authors declare that this submission is in accordance with the principles laid down by the Responsible Research Publication Position Statements as developed at the 2nd World Conference on Research Integrity in Singapore, 2010. Ethical clearance (certificate number M190753) was obtained through the Human Research Ethics Committee (Medical) of the University of Witwatersrand, following signed written consent from the patient's parent for use of the clinical history, clinical photographs, histological tissue sections, photomicrographs, diagnosis and treatment plan.
Signed written consent from the patient's mother for use of his clinical history, clinical photographs, histological tissue sections, photomicrographs, diagnosis and treatment plan were obtained for use in this case report.
All procedures were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008.
Acknowledgements
The authors thank Dr E Ncube for his assistance with the MRI report. Colleagues within the Department of Anatomical Pathology are acknowledged for their agreement of our diagnosis of this unusual case.
Declaration
The authors declare authorship of this article and that they have followed sound scientific research practice. This research is original and does not transgress plagiarism policies.
Author contributions
RW: Reviewed the histology slides, took photomicrographs of the histology slides, contributed to write-up and revision of the manuscript
ZL: Took the clinical photographs, provided the radiograph images and their descriptions, contributed to the clinical write-up of the manuscript
AI: Reviewed the histology slides, contributed to write-up of the manuscript, assisted with photomicrographs of the histology slides
ORCID
Wadee R https://orcid.org/0000-0002-5981-4450
Ismail A https://orcid.org/0000-0001-8635-7630
References
1. Agaimy A, Michal M, Chiosea S, et al. Phosphaturic mesenchymal tumors: clinicopathologic, immunohistochemical and molecular analysis of 22 cases expanding their morphologic and immunophenotypic spectrum. Am J Surg Pathol. 2017 Oct;41(10):1371-80. [ Links ]
2. Yavropoulou MP, Poulios C, Foroulis C, et al. Distant lung metastases caused by a histologically benign phosphaturic mesenchymal tumor. Endocrinol Diabetes Metab Case Rep. 2018;2018:18-0023. https://doi.org/10.1530/EDM-18-0023. [ Links ]
3. Ghorbani-Aghbolaghi A, Darrow MA, Wang T. Phosphaturic mesenchymal tumor (PMT): Exceptionally rare disease, yet crucial not to miss. Autopsy Case Rep. 2017;7(3):32-37. [ Links ]
4. Shiba E, Matsuyama A, Shibuya R, et al. Immunohistochemical and molecular detection of the expression of FGF23 in phosphaturic mesenchymal tumors including the non-phosphaturic variant. Diagn Pathol. 2016 Dec;11(1):26. [ Links ]
5. Bahrami A, Weiss SW, Montgomery E, et al. RT-PCR analysis for FGF23 using paraffin sections in the diagnosis of phosphaturic mesenchymal tumors with and without known tumor induced osteomalacia. Am J Surg Pathol [Internet]. 2009;33(9):1348-54. Available from: https://journals.lww.com/ajsp/Fulltext/2009/09000/RT_PCR_Analysis_for_FGF23_Using_Paraffin_Sections.11.aspx. [ Links ]
6. Folpe AL. Phosphaturic mesenchymal tumors: A review and update. Paraneoplastic Syndr. 2019 Jul 1;36(4):260-68. [ Links ]
7. Folpe AL. Phosphaturic mesenchymal tumour. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: IARC Press; 2013. p. 211-12. [ Links ]
8. Wu H, Bui MM, Zhou L, et al. Phosphaturic mesenchymal tumor with an admixture of epithelial and mesenchymal elements in the jaws: clinicopathological and immunohistochemical analysis of 22 cases with literature review. Mod Pathol. 2019 Feb;32(2):189-204. [ Links ]
9. Carter JM, Caron BL, Dogan A, Folpe AL. A novel chromogenic in situ hybridization assay for FGF23 mRNA in phosphaturic mesenchymal tumors. Am J Surg Pathol. 2015 Jan;39(1):75-83. [ Links ]
10. Chande S, Bergwitz C. Role of phosphate sensing in bone and mineral metabolism. Nat Rev Endocrinol. 2018 Nov;14(11):637-55. [ Links ]
11. Sent-Doux KN, Mackinnon C, Lee J-C, Folpe AL, Habeeb O. Phosphaturic mesenchymal tumor without osteomalacia: additional confirmation of the 'nonphosphaturic' variant, with emphasis on the roles of FGF23 chromogenic in situ hybridization and FN1-FGFR1 fluorescence in situ hybridization. Hum Pathol. 2018 Oct 1;80:94-98. [ Links ]
12. Lee J-C, Su S-Y, Changou CA, et al. Characterization of FN1-FGFR1 and novel FN1-FGF1 fusion genes in a large series of phosphaturic mesenchymal tumors. Mod Pathol. 2016 Jul 22;29:1335-46. [ Links ]
13. Honda R, Kawabata Y, Ito S, Kikuchi F. Phosphaturic mesenchymal tumor, mixed connective tissue type, non-phosphaturic variant: Report of a case and review of 32 cases from the Japanese published work. J Dermatol. 2014 Sep;41(9):845-49. [ Links ]
14. Qari H, Hamao-Sakamoto A, Fuselier C, et al. Phosphaturic mesenchymal tumor: 2 new oral cases and review of 53 cases in the head and neck. Head Neck Pathol. 2015 Nov 14;10(2):192-200. [ Links ]
Received: April 2020
Accepted: August 2020
Published: August 2021
* Corresponding author: reubinawadee@gmail.com
Editor: Dr Thomas Hilton, University of Cape Town, Cape Town, South Africa
Funding: No funding was required for this case report.
Conflict of interest: The authors have no conflicts of interest to declare.


{kind=link}
{kind=link}