










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSA Orthopaedic Journal
On-line version ISSN 2309-8309
Print version ISSN 1681-150X
SA orthop. j. vol.14 n.4 Centurion Oct./Nov. 2015
http://dx.doi.org/10.17159/2309-8309/2015/v14n410
TUMOUR
Rosai-Dorfman disease of the distal radius in a child: A case report and review of the literature
Dr MN RasoolI; Dr V GenzenganeII
IMBChB, FCS Orth(SA), PhD(UKZN); Consultant Orthopaedic Surgeon
IIMBChB Registrar Orthopaedics
ABSTRACT
Rosai-Dorfman disease (RDD) is a non-neoplastic self-limiting disease of bone marrow stem cell origin characterised by cervical lymphadenopathy. Primary osseous lesions are rare and the condition can mimic various solitary bone lesions radiologically. A 15-month-old child presented with an isolated, well-defined, lucent lesion of the distal radius. Histology demonstrated numerous large histiocytes with intracytoplasmic lymphocytes, plasma cells and neutrophils. Immunohistochemistry showed CD 68 immunopositivity, confirming RDD. Healing of the lesion was seen 6 months post-operatively following curettage. Isolated extranodal osseous lesions are very rare in children and can mimic several osseous conditions.
Key words: Rosai-Dorfman disease, distal radius, emperipolesis
Introduction
Rosai-Dorfman disease (RDD) is a non-neoplastic, self-limiting disease of bone marrow stem cell origin. It is characterised by bilateral painless cervical lymphadenopathy accompanied by fever, leucocytosis, elevated ESR and hypergammaglobulinaemia.1 The disease has a predilection for the head and neck including the nasal, oral cavities and paranasal sinuses.2 Although extranodal involvement can occur, primary osseous lesions without lymphadenopathy are rare, <2%. It typically affects young adults and adolescents and has a high incidence in peo3ple of African descent.2 RDD is also known as sinus histiocytosis with massive lymphadenopathy (SHML).
Clinically and radiologically the condition can mimic various solitary bone lesions including tumours and infection.3,4 A primary lesion in the right distal radius in a 15-month-old child is reported.
Case report
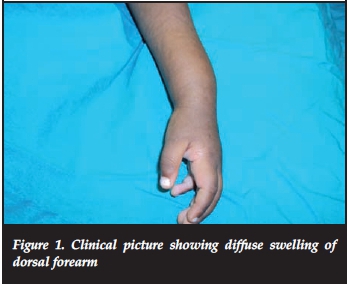
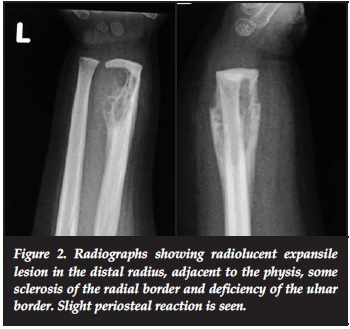
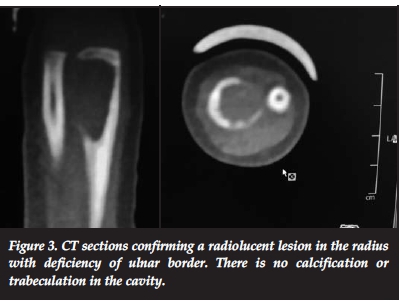
A 15-month-old girl presented with pain and swelling of the distal forearm of 3 months' duration. Clinical examination revealed bony swelling of the distal radius (Figure 1). There was no fluctuance, slight tenderness on palpation and no cervical or axillary nodes. Radiologically a well-defined lucent intramedullary lesion, slightly expansile adjacent to the physis, 5 cm χ 3 cm, was seen with slight periosteal reaction. The radial margin was sclerosed and ulna margin eroded (Figure 2). The Hb was 9 g/dl, WCC 13.0 χ 109/L, ESR 20 mm/hr and CRP <5. The differential count and protein electrophoresis were normal. The Mantoux test was negative. CT scan showed a lucent lesion with scalloping and a thin rim. There was no calcification or fluid in the matrix (Figure 3).
It typically affects young adults and adolescents and has a high incidence in people of African descent
Surgical exploration from a dorsal approach revealed a cavity filled with tissue resembling a caseating granulomatous lesion similar to tuberculosis, extending into the volar aspect of the radius. The cavity was well demarcated; the ulna border was deficient.
Histology demonstrated granulomatous tissue with a mixed inflammatory cell infiltrate composed of plasma cells, lymphocytes, neutrophils and numerous large histiocytes which demonstrated 'emperipolesis' (intracy-toplasmic lymphocytes, plasma cells and neutrophils). The histiocytes showed S-100 and CD-68 immunopositivity. No evidence of tuberculosis or malignancy was seen. Cultures for pyogenic infection were negative. The features were consistent with Rosai-Dorfman disease. Post-operatively the patient recovered well. At 6-months' follow-up, radiographs showed healing of the cavity (Figure 4).

Surgical exploration from a dorsal approach revealed a cavity filled with tissue resembling a caseating granulomatous lesion similar to tuberculosis
Discussion
The histiocytoses are a group of haematologic disorders defined by the pathologic infiltration of normal tissues by cells of the mononuclear phagocytic system. The pathogenic cells central to the development of the histiocytosis arise from a common haematopoetic progenitor. The central cell of this system, the mononuclear phagocyte or histiocyte, represents a group of cells arising from a common precursor, the haematopoetic stem cell.
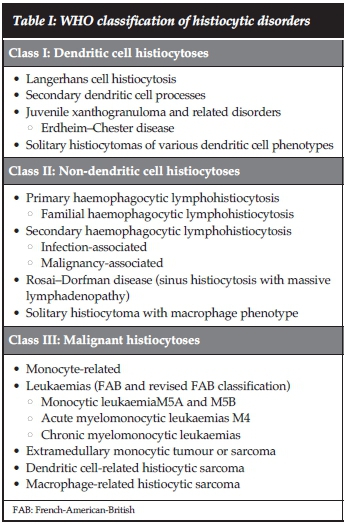
Mononuclear phagocytes can be divided into two major classes, macrophages and dendritic cells. The World Health Organisation classified histiocytic disorders into three classes based on pathologic cells present within the lesions namely, dendritic cell histiocytoses, non-dendritic cell histiocytoses and malignant histiocytoses (Table I).
Langerhans cell histiocytosis (LCH) is the most common of the dendritic cell-related histiocytic disorders. LCH includes the previously identified disorders known as eosinophilic granuloma, Letterer-Siwe disease and Hand-Schuller-Christian disease.
Sinus histiocytosis with massive lymphadenopathy (SHML) also known as Rosai-Dorfman disease (RDD), is the most common type in the class II group. It is characterised by a lymphohistiocytic accumulation in the sinuses of the lymph nodes.5
Rosai-Dorfman disease is a benign disorder of histio-cytic proliferation of unknown cause, initially described by Rosai and Dorfman in 1969.1 It is a non-neoplastic systemic disease of bone marrow stem cell origin which commonly presents with bilateral, non-tender, painless cervical lymphadenopathy which may be accompanied by fever, raised ESR, weight loss, leucocytosis, anaemia and hypergammaglobulinaemia. Other nodes are less frequently involved.1-3
Extranodal RDD involving the bone poses a clinical and radiological challenge. Primary osseous lesions are rarely reported in children. Sundaram reviewed the literature and found seven cases of primary osseous involvement in children <17 years of age. The lesions were mainly lytic or cystic in appearance with cortical erosions. Periosteal reaction was uncommon.
Extranodal RDD occurs in 43% of patients with 23% experiencing isolated extranodal disease, commonly involving the head and trunk.2-4 Osseous involvement with nodal disease is rare <8%; most of these consist of multiple lesions involving the same bone. Primary solitary osseous manifestation without lymphadenopathy is even more rare accounting for <2% of all reported cases.6-8
The aetiology of RDD is unknown. It is thought to be genetic or an immunological disorder, postulated to be a response to an infectious agent with subsequent lymph node proliferation.2,8 This in turn leads to an immunosuppressed state as proliferates of histiocytes phagocytose active lymphocytes. It has also been documented with immune disorders.2 Other infectious agents such as herpes virus, Epstein-Barr virus and cytomegalovirus have also been implicated to be associated with RDD.2
The mean age of onset is usually about 20 years.2 In isolated osseous involvement the location of the lesions was mainly in the lower limbs (tibia and femur). Lesions in the hand, spine, facial and skull bones have been reported. In children distal radius involvement has been reported on two occasions only.6,9 Pain and swelling were the main features. The lesions were part of a multifocal involvement.
Radiographs commonly show a lytic medullary lesion with a sclerotic border. There may be cortical erosion and periosteal reaction.5 A bone scan is useful to show other areas of increased uptake. CT and MRI scans may help with evaluation of the extent of the lesion, the contents of the lesion and any fluid levels in the cystic cavities.
The radiological and clinical features of RDD can mimic various common osseous lesions and the diagnosis can be delayed. Simple and aneurysmal bone cysts, tuberculosis, subacute osteomyelitis, Langerhan's cell histiocytosis (LCH), chondroblastoma, non-ossifying fibroma, monostotic fibrous dysplasia, lymphoma, leukaemia and metastases have been considered by some authors in children.3,7-11 All these conditions have variable presentations clinically and radiologically which makes the diagnosis more difficult.
In the literature Langerhans cell histiocytosis (LCH) is the most closely related condition to RDD based on radiological findings. However LCH differs from RDD based on histo-logical presentation. LCH usually exhibits many eosinophils and histiocytes. Immuno-histochemical staining is always CD1a positive in LCH. Eosinophils are absent in RDD and CD-68 immunopositivity is the distinguishing feature.
Radiographs commonly show a lytic medullary lesion with a sclerotic border. There may be cortical erosion and periosteal reaction
The diagnosis is confirmed on characteristic histological features of 'emperipolesis', i.e. histiocytic phagocytosis, which is characterised by intact inflammatory cells in the cytoplasm of large histiocytes in RDD. This is an important clue in the pathological diagnosis. Although the typically described intracytoplasmic inflammatory cells are lymphocytes, other cells such as plasma cells, erythro-cytes and polymorphonuclear leucocytes can be present.
In our environment cystic tuberculosis, subacute osteomyelitis and fungal infections are more common conditions that can resemble RDD.7 Primary lymphoma of bone can present with destructive bone lesions, typically B cell non-Hodgkin's variety. This tumour is becoming more prevalent in our environment due to HIV infection.12 Nephroblastoma is the commonest primary tumour with metastases to bone.
The clinical course of RDD is typically self-limiting. Greater than 70% have spontaneous resolution.1,2,10,11 A small subset relapse. The prognosis is worse with disseminated involvement of nodal sites. No malignant transformations have been reported. Studies that have focused on skeletal manifestations have documented success with curettage or surgical resection.13 Bone graft was used to fill defects of the talus in one report.3 Steroid administration induces the disappearance of fever and reduction of lymph node size in patients with soft tissue involvement.14 There is no information on its use in preventing osseous disease. Extraosseous and intraosseous lesions can develop after a disease-free interval.1,8 Although response to chemotherapy has been documented, including a combination of alkylating agents with vincristine and steroids and the combination of methotrexate and vincristine, chemotherapy has been shown to be ineffective.15 Radiotherapy has limited efficacy, but occasional response may occur.15 Further data on the use of interferon is needed to define the role of immunomodulators in RDD.
In conclusion, extranodal primary osseous presentation of RDD may present a diagnostic challenge. The condition must be included in the differential diagnosis of lytic or lucent lesions of the skeleton.
References
1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy a newly recognized benign clinico- pathological entity. Arch Pathol. 1969;87:63-70. [ Links ]
2. Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73. [ Links ]
3. Allegranza A, Barbareschi M, Solero CL, et al. Primary lymphohistiocytic tumour of bone: a primary osseous localization of Rosai-Dorfman disease. Histopathology. 1991;18:83-86. [ Links ]
4. Walker PD, Rosai J, Dorfman RF. The osseous manifestations of sinus histiocytosis with massive lymphadenopathy. Am J Clin Pathol. 1981;75:131-39. [ Links ]
5. Shahlaee AH, Arceci RJ. Histiocytic disorders. In Arceci RJ, Hann IM, Smith OP (eds). Paediatric Haematology (3rd edition). Massachusetts: Blackwell Publishing; 2006:693-723. [ Links ]
6. Sundaram C, Uppin SG, Chandrashekar P, et al. Multifocal osseous involvement as the sole manifestation of Rosai- Dorfman disease. Skeletal Radiol. 2005;34:658-64. [ Links ]
7. Rasool MN, Ramdial PK. Osseous localization of Rosai-Dorfman disease. J Hand Surg (Br.) 1996;21:349-50. [ Links ]
8. Kang RW, McGill KC, Lin J, Gitelis S. Chronic ankle pain and swelling in a 25 year old woman. Clin Orthop Relat Res. 2011;469:1517-21 [ Links ]
9. Rodriguez-Galindo C, Helton KJ, Sanchez ND, et al. Extranodal Rosai-Dorfman disease in children. J Pediatr Hematol Oncol. 2004;26:19-24. [ Links ]
10. Dean EM, Wittig JC, Vilalobos C, Garcia RA. A 16 year old boy with multifocal, painless osseous lesions. Clin Ortho Rel Res. 2012;470:2640-45. [ Links ]
11. Loh SY, Tan KB, Wong YS, et al. Rosai-Dorfman disease of the triquetrum without lymphadenopathy. A case report. J Bone Joint + Surg. 2004;86-A:595-98. [ Links ]
12. Opie J. Haematological complications of HIV infection. S Afr Med J 2012;102:465-68. [ Links ]
13. Walczak BE, Halperin DM, Bdeir RW and Irwin RB. A 50- year-old woman with persistent knee pain. Clin Orthop Rel Res. 2011;469:3527-32. [ Links ]
14. Oka M, Kamo T, Goto N, et al. Successful treatment of Rosai-Dorfman disease with low-dose oral corticosteroid. J Dermatol. 2009;36:237-40. [ Links ]
15. Pulsoni A, Anghel G, Falcucci P, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case and literature review. Am J Hematol. 2002;69:67-71. [ Links ]
 Correspondence:
Correspondence:
Dr MN Rasool
Department of Orthopaedic Surgery
University of KwaZulu-Natal Nelson R Mandela School of Medicine
Private Bag 7 Congella, Durban, 4001
Tel: 031-2604297
Email: rasool@ukzn.ac.za
No benefits of any form have been or are to be received from a commercial party related directly or indirectly to the subject of the article

