










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Orthopaedic Journal
versão On-line ISSN 2309-8309
versão impressa ISSN 1681-150X
SA orthop. j. vol.14 no.4 Centurion Out./Nov. 2015
http://dx.doi.org/10.17159/2309-8309/2015/v14n4a1
BONE METABOLISM
Cell signalling and bone remodelling: The skeleton as an endocrine relay organ Part 1
EJ RaubenheimerI; HD HendrikII
IPhD, DSc ; Metabolic Bone Disease Unit, Faculty of Health Sciences, SMU, South Africa
IIMSc; Metabolic Bone Disease Unit, Faculty of Health Sciences, SMU, South Africa
ABSTRACT
As knowledge on the signalling pathways involved in bone remodelling unfolds, maintenance of skeletal health and the management of skeletal diseases will increasingly focus on the manipulation of the autocrine, paracrine and endocrine mechanisms involved in the process. This overview is aimed at providing practitioners with an update on recent advances on cell signalling in bone remodelling and highlights the role of the skeleton in systemic metabolism.
Key words: bone metabolism, cell signalling, skeletal remodelling
Introduction
Until recently the functions of the skeleton were believed to be limited to the maintenance of the vital ionised blood calcium (Ca2+) concentration, provision of rigid attachments for muscles, protection of vital organs and hosting of the haemopoietic system. Mapping of the human genome paved the way for the study of rare genetic skeletal diseases, which uncovered signalling pathways involved in bone remodelling. Several of these pathways link systemic health to bone metabolism and the skeleton is now firmly established as an endocrine relay organ. The preventative and corrective management of generalised skeletal deficiency states will increasingly be oriented towards manipulation of cell signalling and, depending on the desired clinical outcome, it is now possible to control the induction or inhibition of bone formation. Space does not permit a detailed review and readers are referred to the voluminous literature on each cytokine mentioned in this manuscript for more detail. This overview is aimed at providing practitioners with insight into cell signalling during bone remodelling and the systemic metabolic implications thereof.
Bone remodelling
The skeleton undergoes remodelling throughout life. The cycle involves resorption of bone through osteoclast action and the substitution of resorbed bone through osteoblast action, resulting in replacement of the entire skeleton every decade.1 In broad terms, this process is aimed at the stabilisation of blood calcium (Ca2+), repair of damaged bone, removal of old bone and enabling the skeleton to meet changing mechanical demands. During the last decade genetic technology unravelled the pathogenesis of several metabolic bone diseases, and important systemic pathways linked to bone were uncovered. This added significantly to our understanding of the role bone plays in systemic health. The homing of precursor cells to sites of bone remodelling, their differentiation and actions are tightly controlled through autocrine, paracrine and endocrine mechanisms mediating an effective and coordinated cycle of resorption, followed by bone formation.
During the last decade genetic technology unravelled the pathogenesis of several metabolic bone diseases, and important systemic pathways linked to bone were uncovered
The sites at which this occurs are referred to as bone metabolic units or BMUs. Although not yet clearly understood, it is suggested that osteocytes in the early stage of programmed cell death release cytokines which stimulate the initiation of a BMU.2 This is a feasible explanation for the initiation of bone remodelling along fracture lines or at sites of bone necrosis where osteocytes invariably lose their vitality.
The bone remodelling compartment (or BRC)
The first morphological evidence of a developing BMU is elevation of the cells lining the bone surface and the creation of a canopy which separates the bone marrow from the BMU. Through the release of angiopoietin3 and vascular endothelial growth factor (VEGF),4 a dedicated blood supply is established and monocytes and lymphocytes (supplied by the blood) and undifferentiated mesenchymal cells (which are concentrated around the blood vessel5) enter the site of remodelling, which is now referred to as a bone remodelling compartment or BRC2(Figure 1). Among other factors, parathyroid hormone (PTH) plays an important role in attracting osteoclast precursors to the BRC either through stimulation of the production of sphinosine-1-phosphate (S1P), a potent chemo-attractant for monocytes produced by resident osteoclasts6 or elaboration of the inflammatory cytokine, macrophage colony stimulating factor (M-CSF) by T-lymphocytes.7
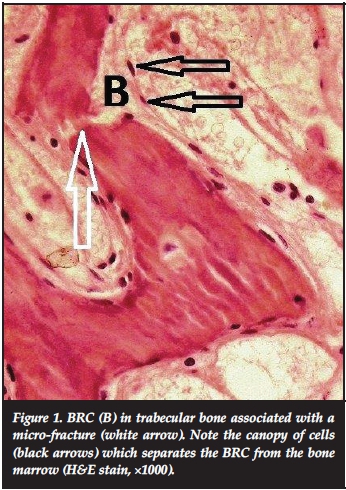
The structure of a BRC in cortical bone differs from that in trabecular bone, which provides a partial explanation for the dissimilar reactions of these two bone compartments to metabolic demands. Trabecular bone is remodelled on its surface, whereas cortical bone remodelling occurs sub-periosteally and within the Haversian systems.
Paracrine and autocrine control of the BMU
Differentiation of monocytes into osteoclasts and undiffer-entiated mesenchymal cells into osteoblasts within a BRC are linked and tightly controlled. Osteoclasts are subjected to paracrine signalling by osteoblasts and are active for approximately 3 weeks.2 One of the most elaborately studied effectors of osteoclast differentiation and activation is RANK-ligand (RANKL) which is produced by osteoblasts. This binds to RANK receptors on the cell surface of osteoclast precursors to activate cytoplasmic kinases which neutralise the inhibition of nuclear factor kB (NF-kB). NF-kB regulates the genes for osteoclast differentiation.8 This signal requires interleukin-1 (IL-1), produced by osteoblasts, for activation.9 The process is balanced by osteoprotegerin (OPG) production by osteoblasts. OPG acts as a decoy receptor for RANKL antagonising its osteo-clastogenic effect.10 Other factors released by osteoblasts and which have an effect on osteoclasts, include parathyroid hormone related protein (PTHrP) and interleukin-1 (IL-1). The latter was previously known as osteoclast activation factor (OAF). PTHrP binds to the same receptors as PTH, activates osteoclasts and mobilises skeletal Ca2*.11 IL-1 enhances osteoclast formation and survival, RANKL expression by osteoblasts and induces IL-6 production in the presence of 1,25(OH)2vit D (vit D3). IL-6 is an inflammatory cytokine known for its resorptive effect on bone.12
For resorption to take place, osteoclasts must bind to specific receptors on the bone surface. 25(OH) vit D (vit D2) reduces the resorptive capacity of osteoclasts by affecting their adhesive properties to bone.13 The specialised ruffled cytoplasmic borders of osteoclasts enlarge the contact area with bone, and the lowered pH, established through the release of H+ ions, creates an acidic environment that is conducive to the release of minerals. The collagen matrix is degraded through the release of cathepsin K and tartrate-resistant acid phosphatases (TRAP).14 Several proteins with systemic metabolic effects are released during bone resorption, establishing an important role for the skeleton in systemic metabolism. The microscopic manifestation of an active site of bone resorption is a resorptive cavity (or Howship's lacuna) containing osteoclasts (Figure 2).
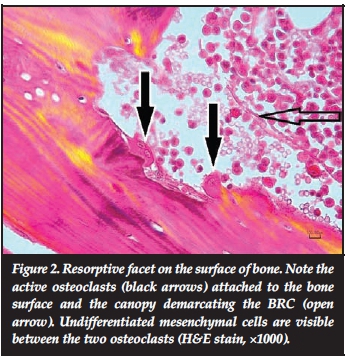
For resorption to take place, osteoclasts must bind to specific receptors on the bone surface
Undifferentiated mesenchymal cells, which are progenitors for several mesenchymal cell types, are programmed to differentiate into osteoblasts through the binding of Wnt ligands to cell-surface Wnt receptors (frizzled family receptors and co-receptor LRP5).15 The cytoplasmic reaction resulting from Wnt triggering in the mesenchymal stem cells can be ß-catenin dependent (canononical pathway) or ß-catenin independent (non-cananonical pathway). In the canononical pathway, several cytoplasmic kinases which neutralise ß-catenin preventing its nuclear translocation, are inactivated. The cytoplasmic concentration of ß-catenin subsequently increases promoting the nuclear signal for osteoblast differentiation and bone formation. Wnt ligands include members of the transforming growth factor β (TGF-ß) family (bone morphogenetic proteins BMP7, BMP6 and BMP3),16 cardiotrophin-1 (produced by osteoclasts)6 and BMP2. BMP7 (also known as osteogenic protein-1) plays a key role in the differentiation of mesenchymal cells into osteoblasts through the phosphorylation of SMAD1 and SMAD5, which activate the transcription of osteogenic genes.17 Wnt antagonists like Dickkopf (Dkkl), Sostdcl and sclerostin are produced by osteocytes and suppress osteoblast function through blocking of the Wnt receptors.18-20 BMP2 is produced by osteoblasts and signals through an autocrine loop for the elaboration of alkaline phosphatase which hydrolyses pyrophosphate (an inhibitor of mineralisation) into phosphate (a promoter of mineralisation).21 Several steps in the Wnt pathway are unique to bone and are currently exploited by industry for the production of monoclonal antibodies and drugs that guide bone remodelling in order to achieve a desirable bone phenotype (see Part 2 - to be published in the SA Orthopaedic Journal Autumn 2016 Vol 15 No 1). Carboxylated osteocalcin released by osteoblasts increases the affinity of the protein matrix for minerals.22 Bone sialoprotein production by osteo-clasts is facilitated by mechanical stress and stimulates osteoblast-induced growth of the hydroxyapatite crystal.22 Osteopontin, a non-collagenous protein deposited in the matrix of bone by osteoblasts, provides a direct mechanical link (through the cell surface receptor integrin) with the cytoskeleton of bone cells. Mechano-transduction through the integrin receptor plays a role in the maintenance of bone mass during skeletal loading23 and is an important consider ation in disuse osteopaenia.
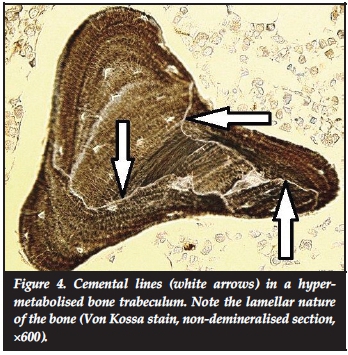
The activities of osteoblasts last for 3-4 months2 and after deposition of immature (woven) bone, which restores the resorbtive facet (Figure 3), the majority undergo apoptosis and those that are incorporated in newly formed bone become osteocytes. Slender processes communicate between neighbouring osteocytes and osteoblasts. This provides a cellular network involved in post formation mineral exchange, transduction of mechanical forces and lamellation of the newly formed bone. The latter mechanism remains one of the enigmas of bone maturation. Irregular cemental lines in lamellar bone, imparting a jigsaw puzzle appearance microscopically (Figure 4), are the only historical record of a restored BRC in mature bone.

Endocrine control of bone remodelling
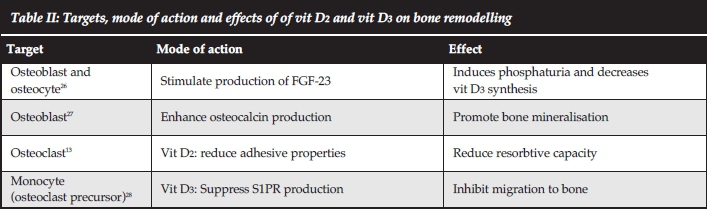
Most of the endocrine factors influencing bone remodelling were described centuries ago. The effects of PTH on the BRC are summarised in Table I and vit D (which is more aptly described as a hormone) in Table II. Prostaglandins E2 and E4, which are widely produced in the body, bind to receptors on the osteoblast, stimulate bone formation and improve fracture healing.29 Nuclear factor kB (NFkB), the 'ageing' hormone produced by the hypothalamus, stimulates the differentiation of osteoclasts and is partially responsible for the reduction of skeletal mass during ageing.30 Epidermal growth factor (EGF) binds to a receptor on osteoblasts and stimulates bone formation.31
Nuclear factor kB (NFkB), the 'ageing' hormone produced by the hypothalamus is partially responsible for the reduction of skeletal mass during ageing
The role of thyroidal calcitonin (TCT) in bone remodelling remains speculative.32 Oestrogen binds to estrogen receptor alpha (ERa) on cortical osteoblasts and maintains cortical bone volume (possibly through the suspension of sclerostin production). ERa of cortical osteoblast progenitors also stimulate Wnt signalling and cortical bone formation in response to mechanical strain, independent of oestrogen. Androgen receptors on mature osteoblasts facilitate the maintenance of trabecular bone mass in males, but are not required for the anabolic effects of androgen on cortical bone.33 The only endocrinologic functions of PTHrP, produced by lactating mammary glands and the placenta, are mobilisation of skeletal calcium for milk production and regulation of maternal placental Ca2+ transport for foetal skeletal growth respectively.11
The skeleton as an endocrine relay organ
The skeleton is now firmly established as an organ influencing peripheral sugar homeostasis. Insulin produced by pancreatic β-cells inhibits OPG production by osteoblasts thereby effectively promoting resorption of bone. During resorption, the elaboration of osteocalcin (which is decar-boxylated in the acidic environment of active osteoclasts) stimulates the release of pancreatic insulin in a forward-feed loop,34 increases tissue insulin sensitivity and promotes male fertility by stimulating testosterone synthesis by Leydig cells.34 Leptin, a hormone produced by adipocytes, binds to receptors in the sympathetic nervous system and suppresses serotonin production. This suspends the serotonin-associated inhibition of osteoblasts and promotes bone formation,35 providing a feasible explanation for the increased skeletal strength of obese individuals. Fibroblast growth factor 23 (FGF23) released by bone cells and elaborated during bone resorption binds to Klotho-FGF23R1 receptors in the renal tubules, induces phosphaturia and decreases vit D3 synthesis,36 thus regulating Ca2+ and P homeostasis.
Insulin produced by pancreatic β-cells inhibits OPG production by 'blasts thereby effectively promoting resorption of bone
Conclusion
Recent revelations in the signalling pathways of bone remodelling propelled the functions of the skeleton from mechanical ambiguity to a key organ in systemic metabolism. A thorough understanding of the signalling pathways involved in bone remodelling is required in order to understand bone disease and master recent developments in the manipulation of bone health, which are discussed in Part 2.
References
1. Parfitt A. Morphological basis of bone mineral measurements: transient and steady effects of treatment in osteoporosis. Miner Electrolyte Metab 1980;4:273-87. [ Links ]
2. Sims NA, Martin TJ. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. BoneKEy Reports 2014; doi: 10.1038/bonekey.2013.215 [ Links ]
3. Park JH, Song HI, Rho JM et al. Parathyroid hormone (1-34) augments angiopoietin-1 expression in human osteoblast-like cells. Exp Clin Endocrinol Diabetes 2006;114:438-43. [ Links ]
4. Street J, Bao M, deGuzman L et al. Vascular endothelial growth factor stimulates bone repair by promoting angio-genesis and bone turnover. Proc Natl Acad Sci USA 2002;99:9656-61. [ Links ]
5. Sacchetti B, Funari A, Michienzi S et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007;131:324-36. [ Links ]
6. Jilke RL, O'Brien CA, Bartelli SM et al. Continuous elevation of PTH increases the number of osteoblasts via both osteoclast-dependent and -independent mechanisms. J Bone Miner Res 2010;25:2427-37. [ Links ]
7. Tawfeek H, Bedi B, Li J-Y et al. Disruption of PTH receptor 1 in T cells protects against PTH-induced bone loss. Plos One 2010; doi10.1371/journal.pone.0012290 [ Links ]
8. Abu-Amer Y. NF-kB signalling in bone resorption. Osteoporos Int 2013; doi:10.1007/s00198-013-2313-x [ Links ]
9. Lee Y-M, Fujikado N, Manaka H et al. IL-1 plays an important role in the bone metabolism under physiological conditions. Int Immunol 2010;22:805-16. [ Links ]
10. Lacey DL, Timms E, Tan HL et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998;93:165-67. [ Links ]
11. McCauley LK, Martin TJ. Twenty-five years of PTHrP progress: from cancer hormone to multifunctioinal cytokine. J Bone Miner Res 2012;27:1231-39. [ Links ]
12. Walsh MC, Kim N, Kadono Y et al. Osteoimmunology: interplay between the immune system and bone metabolism. Ann Rev Immunol 2006:24:33-63. [ Links ]
13. Kogawa M, Findlay DM, Anderson PH et al. Mutation of osteoclastic migration by metabolism of 25OH-vitamin D. J Steroid Biochen Mol Biol 2013,136:59-61. [ Links ]
14. Hayman AR. Tartrate-resistant acid phosphatase (TRAP) and the osteoclast/immune cell dichotomy. Autoimmunity 2008;41:218-23. [ Links ]
15. Williams BO, Insogna KL. Where Wnts went: the exploding field of Lrp 5 and Lrp 6 signalling in bone. J Bone Miner Res 2009;24:171-78. [ Links ]
16. Chen DI, Zhao M, Mundy G. Bone morphogenetic proteins. Growth Factors 2004;22:233-41. [ Links ]
17. Ito F, Asao H, Sugamura K et al. Promoting bone morpho-genetic protein signalling through negative regulation of inhibitory Smads. EMBO J 2001;20:4132-42. [ Links ]
18. Munroe DG, McGee-Lawrence ME, Ousler MJ et al. Update on Wnt signalling in bone cell biology and disease. Gene 2012;492:1-18. [ Links ]
19. Kawakami M, Okuda H, Tatsumi K. Inhibition of Wnt/ β catenin pathway by Dikkopf-1 affects midfacial morphogenesis in chick embryo. J Biosc Bioeng 2013; doi:10.1016/j.biosc.2013.11.015 [ Links ]
20. Moester MJ, Papapoulis SE, Lowik CW et al. Sclerostin: current knowledge and future perspectives. Calcif Tissue Int 2010;87:99-107. [ Links ]
21. Orimo H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch 2010;77:4-12. [ Links ]
22. Karsenty G, Ferron M. The contribution of bone to whole-organism physiology. Nature 2012;481:314-20. [ Links ]
23. Ishijima M, Tsuji K, Ritting SR et al. Osteopontin is required for mechanical stress-dependent signals to bone marrow cells. J Endocrinol 2007;193:235-43. [ Links ]
24. Weitzmann MN. Review Article. The role of inflam matory cytokines, the RANKL / OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica 2013; doi.org/10.1155/2013/125705 [ Links ]
25. Binghurst FR. PTH receptors and apoptosis in osteocytes. J Musculoskelet Neuronal Interact 2002;2:245-51. [ Links ]
26. Rodriguez M, Lopez I, Munoz J et al. FGF23 and mineral metabolism, implications in CKD-MBD. Nefrologia 2012;32:275-78. [ Links ]
27. Cantore FP, Corrado A, Grano M et al. Osteocalcin synthesis by human osteoblasts from normal and osteoarthritic bone after vitamin D3 stimulation. Clin Rheumatol 2004;23:490-95. [ Links ]
28. Sphinosine-1-phosphate-mediated osteoclast precursor monocyte migration is a critical point of control in antibone-resorptive action of active vitamin D. Proc Natl Acad Sci USA 2013;110:7009-13. [ Links ]
29. Li M, Thompson DD, Paralkar VM. Prostaglandin E2 receptors in bone formation. Int Orthop 2007;31:767-72. [ Links ]
30. Zhang G, Li J, Purkayastha S et al. Hypothalamic programming of systemic ageing involving IKK-[bgr], NF-[kgr]B and GnRH. Nature 497;211-16. [ Links ]
31. Zang X, Tamasi J, Lu X et al. Epidermal growth factor plays a positive role in bone metabolism in vivo. J Bone Miner Res 2011:26: 1022-34. [ Links ]
32. Wookey PJ. A review of calcitonin expression in embryonic, foetal and adult tissues, with a hypothesis on the connection between expression during foetal development and disease. Open Zool J 2009:2:53-61. [ Links ]
33. Manolagas SC, O'Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol 2013;9:699-712. [ Links ]
34. Hinoi E. The sympathetic tone mediates leptin's inhibition of insulin secretion by modulating osteocalcin bioactivity. J Cell Biol 2008:183:1235-42. [ Links ]
35. Lee NK, Sowa H, Hinoi E et al. Endocrine regulation of energy metabolism by the skeleton. Cell 2007;130:456-69. [ Links ]
36. Kuro-o M. Overview of the FGF23-Klotho axis. Pediatr Nephrol 2010;25:583-90. [ Links ]
 Correspondence:
Correspondence:
Prof EJ Raubenheimer
Pathology: Metabolic Bone Disease Unit
Faculty of Health Sciences
0204 SMU
Tel: +27 12 5214838 Fax: +27 12 5215274
Email: ejraub@fox5.co.za
The authors have no conflict of interest to declare, and received no direct funding for the writing of the article.


{kind=link}
{kind=link}