










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSA Orthopaedic Journal
versión On-line ISSN 2309-8309
versión impresa ISSN 1681-150X
SA orthop. j. vol.13 no.3 Centurion sep. 2014
TUMOUR AND INFECTION
Alveolar soft part sarcoma in pregnancy: A case report and review of the literature
Y PillayI; N FerreiraI; LC MaraisIII; A MotalaIV
IMBChB; Tumour, Sepsis and Reconstruction Unit, Department of Orthopaedic Surgery, Grey's Hospital, Nelson R. Mandela School of Medicine, University of KwaZulu-Natal, Pietermaritzburg, South Africa
IBSc, MBChB, HDip Orth(SA), FC Orth(SA), MMed(Orth); Tumour, Sepsis and Reconstruction Unit, Department of Orthopaedic Surgery, Grey's Hospital, Nelson R. Mandela School of Medicine, University of KwaZulu-Natal, Pietermaritzburg, South Africa
IIIMBChB, FCS Orth(SA), MMed(Ortho); Tumour, Sepsis and Reconstruction Unit, Department of Orthopaedic Surgery, Grey's Hospital, Nelson R. Mandela School of Medicine, University of KwaZulu-Natal, Pietermaritzburg, South Africa
IVMBChB(UCT),FCPath(Anat)SA; Department of Anatomical Pathology, National Health Laboratory Service, Grey's Hospital, Pietermaritzburg, South Africa
ABSTRACT
Alveolar soft part sarcoma is a rare tumour that mostly affects young female patients.
Most published literature involves isolated cases or small case series. We report the case of a 23-year-old woman who presented with a fast-growing, isolated alveolar soft part sarcoma of the left calf. She had no co-morbidities and was 22 weeks pregnant at the time of presentation. Immunohistochemical analysis of the tumour revealed it to be positive for progesterone receptors. Wide excision was performed, with no local recurrence or systemic spread at six-month follow-up. The fact that the tumour expressed progesterone receptors, combined with previous reports of disease progression during pregnancy, raises the possibility of a hormonal contribution to the pathogenesis. This raises the possibility for novel treatment strategies and warrants further investigation.
Keywords: alveolar soft part sarcoma, tumour, progesterone receptor, pregnancy.
Introduction
Alveolar soft part sarcoma (ASPS) is an extremely rare tumour, with only approximately 200 cases reported in the literature.1,2 These tumours are unique among soft tissue sarcomas, in that no histological variants have been described.1 They commonly affect the deep soft tissues of young adults and account for 0.5-1% of all soft tissue sarcomas.3,4 Due to its rarity, controversy exists with regard to its cellular origin, line of differentiation and optimal treatment.1,4,5
Adolescents and young adults between the ages of 15 and 35 years are mostly affected. An overall female predominance is observed, although an age-related gender ratio inversion has been reported.2,4,6,7 While the majority of affected individuals before the age of 30 years are female, males are more commonly affected after the age of 30 years.4,6,7
ASPS generally present as a soft, painless, slow-growing mass.1,4 They are typically located in the deep tissues of the buttock and thigh, but any site may be involved.1 Primary tumours have also been reported to originate in the lungs, orbit, bladder and gynaecological tract.2,5,8 Zhang et al. reported 38 cases involving the female genital tract, including the vulva, vagina, cervix, uterus and broad ligament.9
Due to a relatively indolent but relentless course, patients often present with a protracted history and symptoms of metastatic disease.1,7 Metastases to bone, lung, brain and liver have been reported and are detected in 20% of patients at diagnosis.7,10 Reichardt et al. reported a 30% incidence of brain metastases, which is three times higher than any other soft tissue sarcoma.11 This, unfortunately, denotes a very poor prognosis, as patients with brain metastases have a median survival of 12 months.4
Results from the MD Anderson Cancer Center in Texas showed a 71% five-year survival in patients who presented with localised disease, while patients who presented with metastatic disease only had a 20% five-year survival.7 Lieberman reported survival figures from the Memorial Sloan-Kettering Cancer Center in New York: patients who presented without metastases had survival figures of 77% at 2 years, 60% at 5 years, 38% at 10 years and 15% at 20 years.6
Pathology
Macroscopic
Lesions have a yellow-grey appearance with mixed firm and friable areas on macroscopic examination. Despite the lack of a capsule, tumours are well circumscribed. On cross-section, a white-tan surface with grey-red areas of necrosis and haemorrhage is identified (Figure 1).
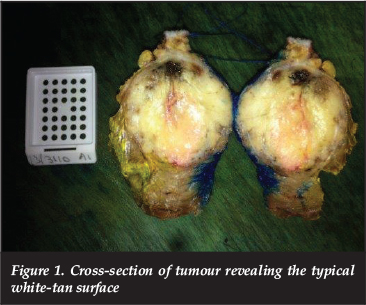
Microscopic
Histological evaluation reveals uniform nests of polygonal cells separated by fibrovascular septae and capillary-sized vascular channels.1,9 Prominent cellular discohesion leads to a distinctive pseudoalveolar pattern.1,4 Cells contain one or more vesicular nuclei with prominent nucleoli, and mitotic figures are rare.1,3,12 The cytoplasm contains abundant eosinophilic granules that are Periodic acid Schiff (PAS) positive and diastase-resistant.1,2,9,13 These granules have been identified as crystals consisting of aggregates of the monocarboxylate transporter protein MCT1 and its cellular chaperone CD147.1,4,14 These intracellular crystalline structures are one of the histological hallmarks of ASPS, in conjunction with the appropriate architectural pattern and cytomorphology.1,3,15 These sarcomas are not usually graded according to conventional tiered grading systems as the neoplasms do not attempt to recapitulate normal histo-anatomic structures and are regarded, by definition, as high-grade sarcomas.
Immunohistochemistry
ASPS cells are positive for non-specific markers such as neuron-specific enolase and vimentin in 30-50% of cases.1 Desmin expression can be demonstrated in 50% of cases. Apart from these non-specific findings, staining for epithelial markers, neuro-endocrine markers and specific melanocytic markers are negative.1
Nuclear expression of TFE3 is a characteristic feature of ASPS. TFE3 is typically expressed in granular cell tumours and in a subset of renal cell carcinomas associated with Xp11 translocations. Ki-67 (a proliferation marker) has been proposed as a potential histologic prognostic marker.16
Due to a relatively indolent but relentless course, patients often present with a protracted history and symptoms of metastatic disease
Electron microscopy
Membrane-bound intracytoplasmic rhomboid crystalline structures are a pathognomonic feature of ASPS, but may not be identified in all cases. Some cases exhibit PAS-positive diastase-resistant granules, which are putative precursors of the crystalline structures.
Cytogenetics
A tumour-specific chromosomal translocation der(17)t(X;17)(p11;q25) has consistently been identified in ASPS cells.17-19 This unbalanced translocation results in the fusion of the Transcription Factor 3 (TFE3) gene at Xp11 to the ASPL gene at 17q25 creating an oncogenic ASPL-TFE3 fusion gene.19,20 Female predominance has been attributed to their double X chromosome genome, leading to a theoretical increased risk for an x-autosome translocation.4,19 This does not, however, explain the age-related gender ratio inversion.
Treatment
The most effective treatment of ASPS remains unclear. Due to the relative rarity of these tumours, large randomised trials have not been conducted. Recurrence rates ranging from 10-31% have been reported,7,19,21,22 but this can be dramatically reduced depending on the treatment strategy.
Ogose et al. and Ogura et al. reported no recurrence in 38 and 18 patients respectively, treated with wide resection without adjuvant radiotherapy.4,23 Anderson et al. and Sherman et al. had no recurrence in 14 and six patients respectively, who were treated with wide resection and radiotherapy, while Ogura reported no recurrence after marginal resection and radiotherapy in two patients.4,24,25 However, Ogose et al. reported recurrence in four out of seven patients who underwent marginal excision without radiotherapy.23 From this data, it appears that wide excision is important for local control. With marginal resection, adjuvant radiotherapy might be necessary to achieve the same result.
Chemotherapy does not appear to provide any additional benefit.1, 4, 10 Ogura et al. treated 26 patients with various conventional chemotherapy treatment combinations with no clinical benefit.4 Most other series came to a similar conclusion, finding ASPS to be resistant to conventional chemotherapy. Various clinical trials are currently underway that target specific tumour characteristics such as the MET receptor tyrosine kinase gene induced by the ASPL-TFE3 fusion protein.10 Three specific trials investigated ArQule, a selective inhibitor of the c-Met receptor tyrosine kinase, sunitinib malate, and sorafenib, both multitargeted tyrosine kinase inhibitors; all three of these drugs show promising results after early testing.10 Other clinical trials using anti-angiogenic agents like bevacizumab and cediranib have also been initiated.
We report the case of a 23-year-old pregnant female patient who presented with a fast-growing ASPS of the calf. Conventional staging and management had to be altered to ensure the safety of both the mother and foetus.
Case report
A 23-year-old female was referred to our tertiary level tumour unit with a six-month history of an enlarging painful mass in the left calf. The patient had no comorbidities and was 22 weeks pregnant. She had initially complained of the mass at a pre-natal visit, and was subsequently referred to the orthopaedic department.
On presentation the patient had a mass, measuring approximately 6 cm in diameter, in the posterior aspect of the left calf. It had poorly discernable edges, was mildly tender, and no overlying skin changes were present. The distal pulses and peripheral nerves were intact.
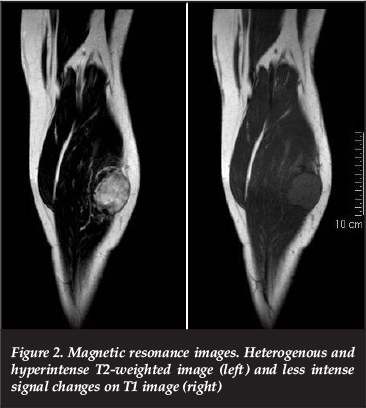
Local staging included radiographs and an MRI scan. The radiographs revealed an increased soft tissue shadow without any bony involvement. MRI scans showed a well-circumscribed lesion arising from the left flexor hallucis longus measuring 6.3 cm x 4.3 cm x 3.4 cm. It appeared confined to the posterior compartment of the leg although peroneus muscle involvement could not be excluded. The lesion appeared heterogeneous and hyperintense on STIR and T2-weighted images. While the tumour exhibited less intense signal changes on the T1 sequence, it was still more intense than the surrounding muscle (Figure 2). There was no involvement of surrounding subcutaneous tissue, bone, nerves or vascular structures. Systemic staging involving computerised tomography (CT) scan of the chest and abdomen was deferred in order to avoid radiation of the foetus.
An incisional biopsy was performed. Microscopic evaluation revealed a tumour composed of nests and alveolar structures lined by cells with enlarged nuclei, prominent nucleoli and abundant dense eosinophilic cytoplasm with PAS-positive diastase resistant intracytoplasmic granules. The cells were separated by fine strands of fibrous tissue, traversed by thin-walled vessels (Figures 3 and 4). Tumour cells expressed vimentin and desmin (focal). Non-specific cytoplasmic myogenin staining was present and as this is a nuclear marker, expression in this case was regarded as negative. CK7, CK20, AE1-AE3, EMA, S-100 and HMB-45 stains were all negative and excluded the histologic differential diagnoses of granular cell tumour, metastatic renal cell carcinoma and metastatic malignant melanoma.
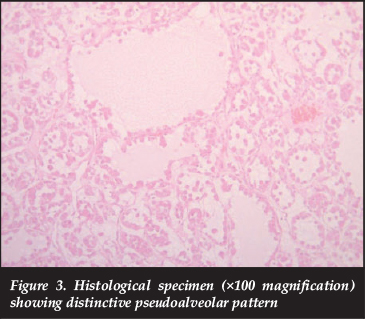
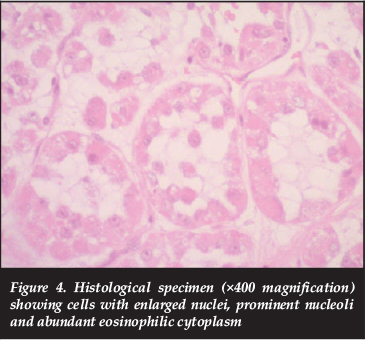
Following histological confirmation of the diagnosis, a multidisciplinary input regarding possible further management was sought. These included orthopaedics, pathology, oncology, anaesthetics and gynaecology/ obstetrics. Treatment options were discussed with the patient, including the potential advantages and disadvantages of each management strategy. The combined decision was made to proceed with wide resection of the tumour without adjuvant radiotherapy or chemotherapy until the birth of the baby.
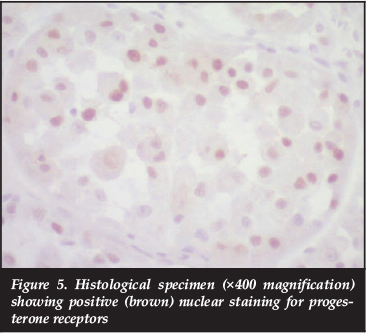
Histological examination of the resected tumour confirmed the initial diagnosis of ASPS. Vascular invasion was identified while evaluation of the resection margins confirmed wide resection on the tumour. In addition, progesterone receptor stain was positive with a positive external control (Figure 5). Oestrogen receptor stain was negative.
Following the delivery of a healthy girl at 35 weeks gestation, systemic staging was completed. A CT scan of the chest and abdomen confirmed the patient to be free of systemic metastases.
Discussion
Cancer during pregnancy is rare, affecting only 1 in 1 000-1 500 pregnancies.26 ASPS is rarer still, accounting for only a fraction of all soft tissue sarcomas. The combination is therefore even more unusual. Only one case has been reported in the literature; Tapisiz, et al. reported a woman diagnosed with lingual ASPS during pregnancy.27 Pregnant patients with malignancy are difficult to manage. Both mother and foetus are at risk. Additionally the usual complete workup and management are complicated. CT scan to diagnose metastatic spread exposes the foetus to excessive and potentially harmful radiation. General anaesthesia for biopsy carries a 1-2% risk of miscarriage and chemotherapy is teratogenic.28 It is important to recognise these potential risks early on and involve a multi-disciplinary team to manage these patients. In our case, the staging CT scan and adjuvant chemotherapy were delayed until after delivery.
Pregnant patients with malignancy are difficult to manage. It is important to recognise potential risks early on and involve a multi-disciplinary team to manage these patients
An interesting finding in this case is the positive progesterone receptor staining. This has been described once previously, in an ASPS case involving the endometrium.29 The authors concluded that the tumour might have originated from endometrial tissue, explaining the finding of progesterone receptors. Most case reports and series in the literature have not specifically evaluated the immunohistochemical expression of hormone receptors in these lesions, even in cases in which lesions developed in sites which contain hormonally responsive tissues. Our patient however had progesterone receptor expression in a lesion arising within skeletal muscle. This might further explain the female predominance in ASPS, and the rapid tumour progression during pregnancy in our patient. Further research into this aspect of ASPS is needed. Most of the tumours previously described in the literature were not associated with pregnancy and the tumour in our case may potentially have been an incidental finding. The presence of hormone receptor expression, however, raises the possibility that this lesion may be related to, or driven by the hormonal milieu related to the underlying gestational state. Hormonal sensitivity was postulated as early as 1988, in a case report describing a patient who developed symptoms related to previously identified lung metastases during pregnancy and the puerperium.30 An additional report from 1960 describes a patient with symptomatic metastases identified during pregnancy, following a pre-pregnancy diagnosis of ASPS.31
Conclusion
Alveolar soft part sarcoma is an extremely rare tumour of mainly young female patients. The current case is no exception to this pattern, but is unusual in its co-existence with pregnancy and its expression of progesterone receptors, which are of potential therapeutic significance in view of the poor response to current conventional chemotherapy. The latter is a direction for further research, as late and ultimately fatal recurrences are a well-recognised phenomenon of ASPS.
Written consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
The authors declare that there is no conflict of interest regarding the publication of this article.
References
1. Folpe AL, Deyrup AT. Alveolar soft-part sarcoma: a review and update. J Clin Pathol. 2006;59(11):1127-32. PubMed PMID: 17071801. Pubmed Central PMCID: 1860509. [ Links ]
2. Mu YL, Liu M, Shi M, Zhao XB, Yin FB, Tang CS, et al. Clinical analysis of alveolar soft-tissue sarcoma of the uterine cervix: a case report. Chin Med J. 2010;123(12):1612-4. PubMed PMID: 20819522. [ Links ]
3. Weiss S. Enzinger and Weiss's Soft tissue tumors. Philadelphia: Mosby; 2008. [ Links ]
4. Ogura K, Beppu Y, Chuman H, Yoshida A, Yamamoto N, Sumi M, et al. Alveolar soft part sarcoma: a single-center 26-patient case series and review of the literature. Sarcoma. 2012;2012:907179. PubMed PMID: 22666000. Pubmed Central PMCID: 3362210. [ Links ]
5. Christopherson WM, Foote FW, Jr., Stewart FW. Alveolar soft-part sarcomas; structurally characteristic tumors of uncertain histogenesis. Cancer. 1952;5(1):100-11. PubMed PMID: 14886902. [ Links ]
6. Lieberman PH, Brennan MF, Kimmel M, Erlandson RA, Garin-Chesa P, Flehinger BY. Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer. 1989;63(1):1-13. PubMed PMID: 2642727. [ Links ]
7. Portera CA, Jr., Ho V, Patel SR, Hunt KK, Feig BW, Respondek PM, et al. Alveolar soft part sarcoma: clinical course and patterns of metastasis in 70 patients treated at a single institution. Cancer. 2001;91(3):585-91. PubMed PMID: 11169942. [ Links ]
8. Amin MB, Patel RM, Oliveira P, Cabrera R, Carneiro V, Preto M, et al. Alveolar soft-part sarcoma of the urinary bladder with urethral recurrence: a unique case with emphasis on differential diagnoses and diagnostic utility of an immunohistochemical panel including TFE3. Am J Surg Pathol. 2006;30(10):1322-25. PubMed PMID: 17001165. [ Links ]
9. Zhang LL, Tang Q, Wang Z, Zhang XS. Alveolar soft part sarcoma of the uterine corpus with pelvic lymph node metastasis: case report and literature review. Int J Clin Exp Pathol ISO. 2012;5(7):715-19. PubMed PMID: 22977670. Pubmed Central PMCID: 3438771. [ Links ]
10. Mitton B, Federman N. Alveolar soft part sarcomas: molecular pathogenesis and implications for novel targeted therapies. Sarcoma. 2012;2012:428789. PubMed PMID: 22566752. Pubmed Central PMCID: 3337503. [ Links ]
11. Reichardt P, Lindner T, Pink D, Thuss-Patience PC, Kretzschmar A, Dorken B. Chemotherapy in alveolar soft part sarcomas. What do we know? Eur J Cancer. 2003;39(11):1511-16. PubMed PMID: 12855256. [ Links ]
12. Rosai J. Rosai and Ackerman's Surgical Pathology. 9th ed. Philadelphia: Mosby; 2004. [ Links ]
13. Ordonez NG. Alveolar soft part sarcoma: a review and update. Adv Anat Pathol ISO. 1999;6(3):125-39. PubMed PMID: 10342010. [ Links ]
14. Ladanyi M, Antonescu CR, Drobnjak M, Baren A, Lui MY, Golde DW, et al. The precrystalline cytoplasmic granules of alveolar soft part sarcoma contain monocarboxylate transporter 1 and CD147. Am J Pathol. 2002;160(4):1215-21. PubMed PMID: 11943706. Pubmed Central PMCID: 1867200. [ Links ]
15. Weiss S, Goldblum J. Enzinger and Weiss's Soft tissue tumours. Philadelphia: Mosby; 2001. [ Links ]
16. Sanjuan X, Sobel ME, Yang J, Merino MJ. Alveolar soft part sarcoma: the role of prognostic markers. Ann Diagn Pathol. 2000;4(3):135-42. PubMed PMID: 10919382. [ Links ]
17. Joyama S, Ueda T, Shimizu K, Kudawara I, Mano M, Funai H, et al. Chromosome rearrangement at 17q25 and xp11.2 in alveolar soft-part sarcoma: A case report and review of the literature. Cancer. 1999;86(7):1246-50. PubMed PMID: 10506710. [ Links ]
18. Heimann P, Devalck C, Debusscher C, Sariban E, Vamos E. Alveolar soft-part sarcoma: further evidence by FISH for the involvement of chromosome band 17q25. Gene Chromosome Cane. 1998;23(2):194-97. PubMed PMID: 9739024. [ Links ]
19. Ladanyi M, Lui MY, Antonescu CR, Krause-Boehm A, Meindl A, Argani P, et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20(1):48-57. PubMed PMID: 11244503. [ Links ]
20. Bu X, Bernstein L. A proposed explanation for female predominance in alveolar soft part sarcoma. Noninactivation of X; autosome translocation fusion gene? Cancer. 2005;103(6):1245-53. PubMed PMID: 15693033. [ Links ]
21. Van Ruth S, van Coevorden F, Peterse JL, Kroon BB. Alveolar soft part sarcoma. a report of 15 cases. Eur J Cancer. 2002;38(10):1324-28. PubMed PMID: 12091061. [ Links ]
22. Evans HL. Alveolar soft-part sarcoma. A study of 13 typical examples and one with a histologically atypical component. Cancer. 1985;55(4):912-17. PubMed PMID: 3967185. [ Links ]
23. Ogose A, Yazawa Y, Ueda T, Hotta T, Kawashima H, Hatano H, et al. Alveolar soft part sarcoma in Japan: multi-institutional study of 57 patients from the Japanese Musculoskeletal Oncology Group. Oncology. 2003;65(1):7-13. PubMed PMID: 12837977. [ Links ]
24. Sherman N, Vavilala M, Pollock R, Romsdahl M, Jaffe N. Radiation therapy for alveolar soft-part sarcoma. Medical and pediatric oncology. 1994;22(6):380-83. PubMed PMID: 7512190. [ Links ]
25. Anderson ME, Hornicek FJ, Gebhardt MC, Raskin KA, Mankin HJ. Alveolar soft part sarcoma: a rare and enigmatic entity. Clin Orthop Relat Res. 2005;438:144-48. PubMed PMID: 16131883. [ Links ]
26. Pentheroudakis G, Pavlidis N. Cancer and pregnancy: poena magna, not anymore. Eur J Cancer. 2006;42(2):126-40. PubMed PMID: 16326099. [ Links ]
27. Tapisiz OL, Gungor T, Ustunyurt E, Ozdal B, Bilge U, Mollamahmutoglu L. An unusual case of lingual alveolar soft part sarcoma during pregnancy. Taiwan J Obstet Gynecol. 2008;47(2):212-14. PubMed PMID: 18603509. [ Links ]
28. Duncan PG, Pope WD, Cohen MM, Greer N. Fetal risk of anesthesia and surgery during pregnancy. Anesthesiology. 1986;64(6):790-94. PubMed PMID: 3717642. [ Links ]
29. Kasashima S, Minato H, Kobayashi M, Ueda Y, Oda Y, Hashimoto S, et al. Alveolar soft part sarcoma of the endometrium with expression of CD10 and hormone receptors. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2007;115(7):861-65. PubMed PMID: 17614855. [ Links ]
30. Pang JA, Yeung TF, Cockram CS. Alveolar soft-part sarcoma: a hormone-sensitive tumour? J Postgrad Med. 1988;64(751):386-88. PubMed PMID: 3200782. Pubmed Central PMCID: 2428676. [ Links ]
31. Farquharson M. Alveolar soft part sarcoma. BMJ. 1960;2(5205):1068-69. PubMed PMID: 13698252. Pubmed Central PMCID: 2097914. [ Links ]
 Correspondence:
Correspondence:
Dr Nando Ferreira
Tumour, Sepsis and Reconstruction Unit
Department of Orthopaedic Surgery
Grey's Hospital
Nelson R. Mandela School of Medicine
University of KwaZulu-Natal
3201 Pietermaritzburg, South Africa
Tel: +27 33 897 3000
Email: Nando.Ferreira@kznhealth.gov.za
This article is also available online on the SAOA website (www.saoa.org.za) and the SciELO website (www.scielo.org.za). Follow the directions on the Contents page of this journal to access it.

