










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Orthopaedic Journal
versão On-line ISSN 2309-8309
versão impressa ISSN 1681-150X
SA orthop. j. vol.13 no.1 Centurion Jan./Mar. 2014
METABOLIC
Chronic kidney disease and the skeleton: Pathogenesis, complications and principles of management
B ThibediI; EJ RaubenheimerII; CEE NoffkeIII
IBCur, MBChB. Nephrology Unit, Department of Internal Medicine, Faculty of Health Sciences, Medunsa Campus, University of Limpopo
IIMChD, PhD, DSc. Metabolic Bone Disease Laboratory, Faculty of Health Sciences, Medunsa Campus, University of Limpopo
IIIMSc. Metabolic Bone Disease Laboratory, Faculty of Health Sciences, Medunsa Campus, University of Limpopo
ABSTRACT
Skeletal and extra-skeletal changes in chronic kidney disease are the result of deteriorating mineral homeostasis with disruption of the concentrations of phosphorous, calcium and circulating hormones. With the improved survival brought about by modern management strategies, early recognition of the prognostic determinants is of paramount importance in improving the morbidity and quality of life of renal patients. The aim of this review is to provide a skeletal perspective on the pathogenesis, radiological appearances, complications and principles of management of patients with chronic kidney disease.
Key words: uraemic osteodystrophy, osteitis fibrosa, hyperparathyroidism, renal failure, osteomalacia.
Introduction
Chronic kidney disease (CKD) has become a major health problem with about one in ten adults affected worldwide.1 As the kidney functions in CKD deteriorate, a progressive disruption of mineral homeostasis leads to skeletal and extraskeletal complications which impact on the quality of life and survival of patients. The skeletal changes, collectively referred to as renal osteodystrophy, received the focus of attention in the past. With the increased life expectancy resulting from improved management strategies, long-term extra-skeletal complications have become important prognostic determinants in the CKD patient. The designation 'CKD-Mineral Bone Disorder' (CKD-MBD) has been introduced to encompass the skeletal and extra-skeletal morbidities in CKD patients.2 The aim of this manuscript is to present orthopaedic surgeons with an overview of CKD-MBD with particular reference to skeletal involvement in the CKD patient.
Mineral homeostasis and bone
Maintenance of serum calcium and phosphate concentrations is dependent on the interaction between vitamin D, fibroblast growth factor 23 (FGF23) and parathyroid hormone (PTH). Activation of vitamin D is a multi-step process beginning with UV irradiation of the skin followed by hydroxylation in the liver, yielding 25-hydroxyvitamin D (25OHD), which is the main form in which vitamin D is stored.3 25OHD is further hydroxylated by the enzyme 1-a-hydroxylase in the kidney as well as in other tissue sites to produce 1,25 dihydroxyvitamin D (1,25(OH)2D), which represents the active metabolite.4 1,25(OH)2D, among its multiple effects on other metabolic activities, facilitates calcium, phosphorous and magnesium uptake in the gastrointestinal tract and retention of calcium in the kidneys.3 Through its involvement in calcium homeostasis, 1,25(OH)2D has a regulatory influence on parathyroid gland function and the release of PTH. If serum calcium concentrations fall, secondary hyperparathyroidism results in the release of PTH which up-regulates osteoclastic activity, thereby elaborating calcium from the skeleton. FGF23 is a bone-derived hormone that influences phosphate concentrations by inhibiting its reabsorption in the proximal tubules of the kidney through a mechanism independent of PTH. In addition, FGF23 has a negative influence on 1,25(OH)2D concentrations by facilitating its catabolism and suppressing the activity of 1-a-hydroxylase.4,5
Pathogenesis of CKD-MBD
A progressive retention of FGF23 in CKD, resulting from a drop in the glomerular filtration rate, has been reported to counteract hyperphosphataemia. The increase in the concentration of FGF23 also contributes to a reduction of the concentration of 1,25(OH)2D, with hypocalcaemia and induction of secondary hyperparathyroidism.5 PTH induces increased bone remodelling which further stimulates the elaboration of FGF23 by bone cells. In the more advanced stages of CKD hydroxylation of 25OHD is progressively impaired due to the loss of functioning renal tissue. The lack of 1,25(OH)2D results in decreased absorption of calcium in the gastrointestinal tract and reduced retention of calcium by the diseased kidneys. The consequence is a further decrease in the serum calcium concentration. Hyperphosphataemia stimulates PTH secretion and elaboration of FGF23.5
The latter initially inhibits PTH secretion but this inhibitory effect is lost with the development of parathyroid hyper-plasia in advancing CKD. The impact on bone is defective mineralisation of osteoid (due to low serum calcium concentrations), up-regulation of bone metabolism with activation of osteoclasts (due to PTH overproduction) resulting in loss of mineralised bone and enhanced release of FGF23. In uraemia the parathyroid glands becomes relatively resistant as evidenced by the high PTH concentrations despite high FGF23 concentrations. Although this mechanism serves as a feasible explanation for depletion of mineralised bone in patients with CKD, care should be taken not to oversimplify the disease process.
The metabolic effects of CKD on the skeleton are compounded by several factors. In patients with prolonged secondary hyperparathyroidism, the parathyroid glands may fail to respond to serum calcium concentrations, and an irreversible phase of PTH overproduction, designated as tertiary hyperparathyroidism, could develop.6 The mechanism involved in the transformation of parathyroid function towards autonomous PTH release is unclear. With tertiary hyperparathyroidism the parathyroid response to conventional management becomes ineffective and may require parathyroidectomy to correct.
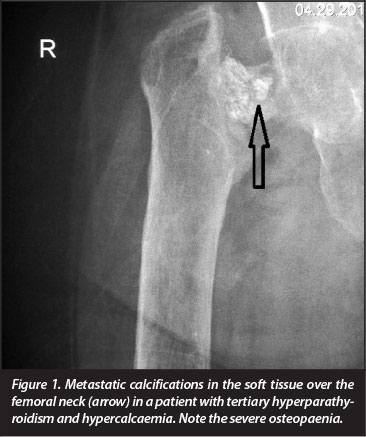
If the release of calcium through bone catabolism results in prolonged hypercalcaemia, progressive soft tissue calcification, also referred to as 'metastatic calcification' occurs (Figure 1). There is a significant correlation between hypercalcaemia and the higher risk of vascular and valvular calcifications in patients on long-term haemodialysis.7 More than half the deaths of patients with end-stage CKD are due to cardiovascular complications and the annual cardiovascular mortality rate is more than ten times that of age-matched non-renal disease patients.8 Calciphylaxis, a rare and life-threatening condition where progressive cutaneous necrosis secondary to cutaneous blood vessel calcification occur, may complicate CKD.9 Long-term administration of corticosteroids, as part of the management regimen of certain chronic kidney diseases as well as in recipients of transplanted kidneys, contributes not only to a reduction of the activities of all cells involved in bone metabolism, but also suppresses intestinal calcium uptake.10
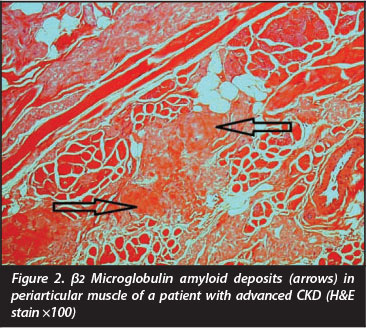
Recent advances in the management of CKD impact positively on the survival of patients. For this reason other chronic complications like β2 microglobulin amyloidosis11 are becoming more pertinent in the literature. The latter results from a reduction of β2 microglobulin excretion, and although modern dialysis modalities allow improved removal of β2 microglobulin, long-term dialysis may contribute to deposits in joints, bone and the surrounding soft tissues12 (Figure 2). The destructive arthropathy and development of subchondral radiolucencies due to the β2 microglobulin amyloid deposits are difficult to distinguish radiologically from brown tumours and it is not surprising that the presence of these deposits has been associated with an increased femoral fracture risk.13,14 β2 microglobulin has recently been reported to stimulate osteoclastogenesis,15 supporting its direct role in bone catabolism in patients with CKD.
Changes in the composition of tissue fluid, resulting from the accumulation of the catabolytes of metabolism due to the reduced glomerular filtration rate of advanced CKD, has a negative impact on metabolic activities of most cells, including those involved in bone metabolism. This together with the changes in volume distribution makes the interpretation of some of the biochemical parameters used to monitor bone metabolism in patients with advanced CKD debatable. For these reasons bone biopsy submitted to a laboratory acquainted with histomorphometric techniques provides superior information on the volumes of mineralised bone, osteoid and bone cell activities in the advanced stages of CKD.16
Microscopic and radiological appearances of renal osteodystrophy
The initial microscopic features of renal osteodystrophy are wide osteoid seams due to a reduced mineralisation rate and increased osteoclastic bone resorption due to elevated PTH. These features are the hallmarks of osteomalacia (or rickets in the growing skeleton).14 With the aid of static and dynamic histomorphometry, which requires tetracycline labelling and special fixation techniques, these changes can be measured and compared to age-matched norms,14-17 providing valuable information on the stage of skeletal catabolism as well the monitoring of the efficiency of a management strategy employed.
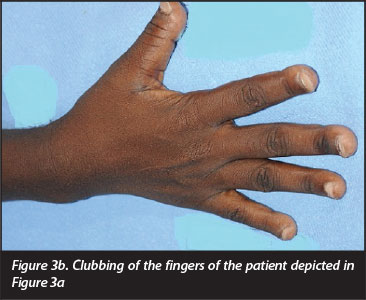
The earliest radiological signs of skeletal involvement are reported to be resorption of the phalanges (Figure 3a and 3b) which starts as sub-periosteal resorption of the radial aspects of the middle phalanges of the hands and sub-endosteal and intra-cortical resorption of long bones. In a more advanced stage, a reduction of the cortical width of long bones and bowing or even fracture of weight-bearing bones occur, and a generalised lack of mineralised bone, with radiological features similar to osteoporosis may develop.16 Vertebral fractures may change the shape of the thoracic cage and sacro-iliac deformities may impair the ability to walk.

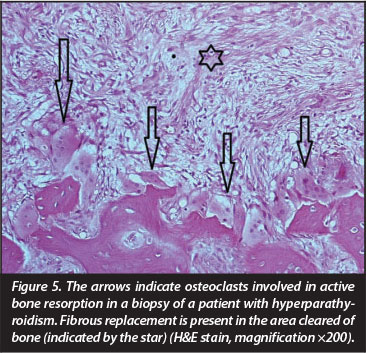
Foci showing fulminant bone resorption with fibrous replacement (Figure 4), referred to as osteitis fibrosa, is the result of surplus PTH due to over-active parathyroid glands. This metabolic state is designated as 'high turnover osteodystrophy'4 and manifests radiologically as well-defined areas of radiolucency often erroneously described as 'osteitis fibrosa cystica' or 'brown tumours of hyperparathyroidism'18 (Figure 5). 'Osteitis fibrosa cystica', a term created by Von Recklinghausen in the nineteenth century19 is neither an 'osteitis' nor 'cystic', but merely the end stage manifestation of PTH-induced fulminant bone resorption. The 'brown tumour of hyperparathyroidism' first described in 195320 is not a 'tumour' in a pathological sense but rather a reparative process. Other sites in the skeleton may show a more diffuse granular appearance also described as a 'salt and pepper' appearance due to trabecular coarsening.18 Bone replacement by fibrous tissue is particularly incapacitating in the growing skeleton. The radiological differential diagnosis of high turnover osteodystrophy in children includes fibrous dysplasia. The former is characterised by its poor corti-comedullary definition, a feature not found in fibrous dysplasia.21 The term 'uraemic leontiasis ossea' was applied in the past to rare cases in which enlargement of the facial bones, due to the high turnover osteodystrophy, was a prominent clinical feature.

In practice the distinction between the different skeletal changes may be difficult as osteomalacia, osteoporosis and high turnover osteodystrophy may occur together in the same patient. Radiological demonstration of pseudofractures (or looser's zones) affecting the scapula, pubic rami and proximal femurs may aid in diagnosing osteomalacia. In a significant number of patients a clear distinction between the skeletal changes is not possible and the term 'mixed bone disease' is used to describe this hybrid manifestation of renal osteodystrophy.6
Adynamic bone disease is a variety of renal osteodystrophy characterised by a complete absence of metabolic activity on bone surfaces, no accumulation of osteoid and low-to-normal serum PTH concentrations.6
This pattern is best diagnosed with a bone biopsy. It is found in a high percentage of patients on dialysis but also in CKD patients treated rigorously with calcium or vitamin D derivatives, diabetics or those who received unnecessary parathyroid surgery. Although the pathogenesis is not clear and many factors may be implicated, it appears as if uraemia contributes to the down-regulation of PTH receptors on bone cells.22
Management of renal osteodystrophy
Addressing the bone changes associated with CKD is complex and it is not within the scope of this paper to provide a detailed account thereof. More information can be obtained from a recently published, evidence-based systematic review published under the auspices of Kidney Disease International Global Outcomes (KDIGO).2 In summary the primary approach in the management should be focused on the restoration of renal function through dialysis or renal transplantation thereby preventing the adverse cardiovascular and skeletal complications of CKD-MBD. From a skeletal perspective, the mainstay of the management includes normalisation of serum calcium, serum phosphate and PTH concentrations. The use of calcimimetic drugs in end-stage renal disease have been shown to reduce the levels of PTH, calcium and phosphate as they mimic the action of calcium.23,24 The manipulation of serum calcium can be supplemented by controlling dietary calcium intake and/or diasylate calcium content. A reduction of dietary phosphate intake contributes towards achieving phosphate homeostasis.6 Administration of vitamin D metabolites (or calcitriol, a 1,25(OH)2D analogue25) corrects the deficiency of 1,25(OH)2D and suppresses secondary hyperparathyroidism, thereby addressing the skeletal morbidity of high turnover osteodystrophy. Novel vitamin D analogues have been developed which may in future have advantages in preventing vascular calcifications resulting from the manipulation of the calcium-phosphate product in the extracellular fluid.26,27 A bone biopsy may provide decisive information as vitamin D metabolites do not improve adynamic renal osteodystrophy.28 Indications for a parathyroidectomy are persistent and non-responsive hypercalcaemia due to tertiary hyperparathyroidism, progressive soft tissue calcification and persistent high turnover osteodystrophy after all strategies of reducing serum calcium concentrations have been exploited.26
Conclusion
An understanding of the pathogenesis of CKD-MBD is essential for preventing the slumbering skeletal and extraskeletal complications in the renal patient. Restoration of serum calcium, phosphate and PTH concentrations not only addresses the catabolism of bone but also plays an important role in improving the mortality rate and quality of life of patients suffering from renal disease.
References
1. Schiepatti A, Remuzzi G. Chronic renal disease as a health problem: epidemiology, social and economic implications. Kidney Int Suppl 2005;68:7-10. [ Links ]
2. Willis K, Cheung M, Slifer S. KDIGO 2012 Clinical practice guidelines for the evaluation and management of chronic kidney disease. Kidney Internat Suppl 2013;1:1-150. [ Links ]
3. Raubenheimer EJ, Noffke CEE. Vitamin D and health - a historical overview. S A Orthopaedic J 2011;10(2):39-43. [ Links ]
4. Al-Badr W, Martin KJ. Vitamin D and kidney disease. Clin J Am Soc Nephrol 2008;3:1555-60. [ Links ]
5. Quarles LD. The role of FGF23 in vitamin D and phosphate metabolism: implications in chronic kidney disease. Exp Cell Res 2012;318:1040-48. [ Links ]
6. Smith R, Wordsworth P, editors. Clinical and biochemical disorders of the skeleton. Oxford: University Press. 2008;191-94. [ Links ]
7. Raggi P, Boulay A, Chasen-Taber S, et al. Cardiac calcification in adult hemodialysis patients. A link between end-stage renal disease and cardiovascular disease. J Am Coll Cardiol 2002;39(4):695-701. [ Links ]
8. Foley RN, Parfrey PS, Samak MJ. Clinical epidemiology of cardiovascular disease. Am J Kidney Dis 1998;32(Suppl):S112-S119. [ Links ]
9. Ivker RA, Woosley J, Briggaman RA. Calciphylaxis in three patients with end-stage renal disease. Arch Dermatol 1995;131(1):63-68. [ Links ]
10. Dovio A, Perazzolo L, Osella G, Ventura M, Termine A, Milano E, Bertolotto A. Immediate fall of bone formation and transient increase of bone resorption in the course of highdose, short term glucocorticoid therapy in young patients with multiple sclerosis. J Clin Endocrinol Metab 2004;89:4923-28. [ Links ]
11. Mount SL, Eliabbakh GH, Hardin NJ, et al. |3-2 microglobulin amyloidosis presenting as bilateral ovarian masses. A case report and review of the literature. Am J Surg Pathol 2002;26:130-33. [ Links ]
12. Kay J, Bardin T. Osteoarticular disorders of renal origin; disease related and iatrogenic. Baillis Clin Rheumatol 2001;14:285-305. [ Links ]
13. Onishi S, Andress DL, Malone NA, et al. Beta-2 microglobulin deposition in bone in chronic renal failure. Kidney Int 1991;39:99-105. [ Links ]
14. Fok WMM, Leung HB. Unresolved lytic lesions following parathyroidectomy in patients with chronic renal failure. J Bone Joint Surg Br 2008;90(4) 506-509. [ Links ]
15. Menaa C, Esser E, Sprague SM. B-2 Microglobulin stimulates osteoclast formation. Internat Soc Nephrol 2008; http://www.kidney-international.org [ Links ]
16. Raubenheimer EJ. Part I: Metabolic bone disease: histomorphometry as a diagnostic tool. S A Orthopaedic J 2008;Spring:19-23. [ Links ]
17. Vigorita VJ. The tissue pathological features of metabolic bone disease. Orthop Clin North Am 1984;15:613-19. [ Links ]
18. Wagener GWW, Sander M, Hough FS. Advanced osteitis fibrosa cystica in the absence of phalangeal sub periosteal resorption. A case report and review of the literature. SAM] 1985;67:31-32. [ Links ]
19. Buchanan WW, Kraag GR, Palmer DG, Cockshott WP. The first recorded case of osteitis fibrosa cystica. Can Med Assoc ] 1981;124:812-15. [ Links ]
20. Campuzano-Zuluaga G, Velasco-Perez W, Martin-Zuluaga JI. A 60-year old man with chronic renal failure and a costal mass: A case report and preview of the literature. ] Med Case Reports 2009;3:7258. [ Links ]
21. Chang JI, Som PM, Lawson W. Unique imageing findings in the facial bones of renal osteodystrophy. Am ] Neuroradiol 2007;28:608-609. [ Links ]
22. Coen G. Adynamic bone disease: an update and an overview. J Nephrol 2005;18:117-22. [ Links ]
23. Zhang Q, Li M, You L, Li H, Ni l, Gu Y, Hao C, Chen J. Effects and safety of calcimimetics in end stage renal disease patients with secondary hyperparathyroidism: a meta-analysis. PLoS ONE 2012;7:e48070. [ Links ]
24. Elder G. Use of calcimimetic drugs. www.medscape.com/ viewarticle/542490. [ Links ]
25. Zhou H, Xu C. Comparison of intermittent intravenous and oral calcitrol in the treatment of secondary hyperparathyroidism in chronic hemodialysis patients: a meta-analysis of randomized control trials. Clin Nephrol 2009;71:276-85. [ Links ]
26. Cozzolino M, Galassi A, Gallieni M, et al. Pathogeneisis and treatment of secondary hyperparathyroidism in dialysis patients: The role of paricalcitrol. Curr Vasc Pharmacol 2008;6(2):148-53. [ Links ]
27. Teng M, Wolf M, Lowrie F, et al. Survival of patients undergoing hemodialysis with paricalcitrol or calcitriol therapy. New Engl J Med 2003;349:446-56. [ Links ]
28. Brandi L, Dougaard H, Nielsen PK, et al. Long term effects of intravenous 1a(OH)D3 combined with CaCo3 and low calcium dialysis on secondary hyperparathyroidism and biochemical bane markers in patients on chronic hemodialysis. Nephron 1996;74:89-103. [ Links ]
 Correspondence:
Correspondence:
Prof EJ Raubenheimer
Room FND 110
Medunsa Campus University of Limpopo
0204 South Africa
Tel: +27 12 5214839
E- mail: Erich.raubenheimer@ul.ac.za

