










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSA Orthopaedic Journal
On-line version ISSN 2309-8309
Print version ISSN 1681-150X
SA orthop. j. vol.12 n.2 Centurion Jan. 2013
BASIC SCIENCE
The Torg-Winchester form of hereditary osteolysis: Orthopaedic manifestations and management
JD BertieI; P BeightonII; D ThompsonIII
IMBChB(Stell) Orthopaedic Medical Officer, Pietermaritzburg Hospital Complex, Pietermaritzburg
IIOMB, MD, PhD, FRCP, FRSSA Emeritus Professor, Division of Human Genetics, Faculty of Health Sciences, University of Cape Town
IIIMBChB(UCT), FRCS(Glas) Principal Specialist Paediatric Orthopaedics, Greys Hospital, Pietermaritzburg
ABSTRACT
The genetic osteolyses are an uncommon group of skeletal disorders in which severe orthopaedic complications can occur. In this article we present the clinical and radiological features of a boy with the Torg-Winchester form of osteolysis and discuss the orthopaedic management of his upper and lower limb deformities. Heritable osteolysis warrants consideration as a possible diagnosis in children presenting with clinical deformity following minimal trauma and radiological evidence of progressive dissolution of bone.
Key words: genetics, orthopaedics, osteolysis, skeletal syndromic
Introduction
The Hereditary Osteolyses are a group of rare genetic disorders characterised by dissolution of the bones and joints.1 As these conditions are progressive and cause considerable physical handicap, ongoing orthopaedic measures are crucial2,3 for effective management.
In the Torg-Winchester form of osteolysis (MIM 259600) the wrists and ankles are mainly affected, with eventual disappearance of the bones of the carpus and tarsus.4 The Nodulosis-Arthropathy-Osteolysis (NAO) syndrome has been incorporated into this disorder, which is now regulated as a spectrum caused by different mutations within the matrix metalloproteinase gene MMP2.57
In view of the rarity of this condition and the paucity of literature pertaining to the orthopaedic management of the osteolyses, we have presented, depicted and discussed our approach to this boy's skeletal problems.
Case report
A Zulu boy aged 6 years presented with complaints of a painful right leg and arm with associated deformity. His development was normal up to the age of 3 years, when a deformity of his right elbow was noticed two months after a fall. At 6 years of age he developed deformities of both hands, as well as the right knee following minimal trauma. He had no family history of similar abnormalities.
He was a healthy looking and intelligent boy, and his weight and height were normal for his age. His spine was normal, and he had no subcutaneous nodules. His musculoskeletal pathology was predominantly right-sided, but the hands and feet were affected bilaterally (Figure 1). He was unable to walk without assistance, due to flexion deformities of the right knee and elbow.


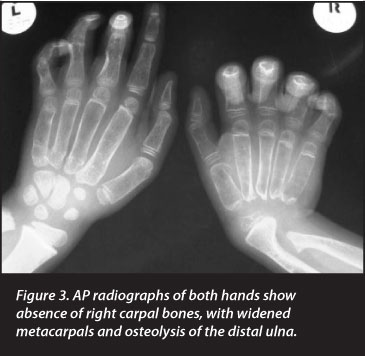

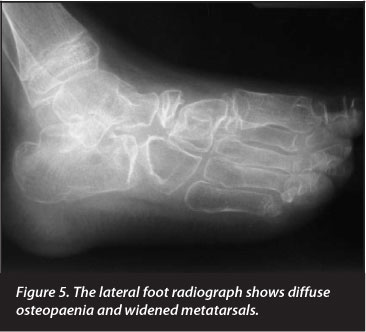

Radiographs revealed generalised skeletal lucency with absence of the carpal bones in the right wrist and destruction of the right elbow joint. The metacarpals, metatarsals and phalanges of both hands and feet were widened, with loss of tubulation. The medial regions of both clavicles were expanded. The pertinent clinical and radiological findings are depicted in Figures 1-6 and listed in Table I.
Routine haematological and biochemical investigations yielded normal results and there was no biochemical evidence of parathyroid dysfunction. His renal function was normal. Facilities for the investigation of the causative MMP2 gene were unavailable.
Orthopaedic management
Management of his upper limb pathology was focused on his right forearm and both hands. We did not consider interfering with his right elbow deformity, as the position would allow him to use his hand to feed himself. Management of the ulna club hand involved a distal ulna osteotomy and wrist spanning external fixator (Orthofix MultiPlanar MiniRail System). The deformity was corrected gradually until the wrist was in a neutral position, and remained in situ for three months. The external fixator was then replaced with a below-elbow cast to prevent recurrence of the deformity, followed by a wrist gaiter. His hands were splinted and exercised from the time of his admission but we were unable to improve his phalangeal flexion contractures.
The management of his lower limb pathology was focused on his right knee flexion contracture. He was managed in a reverse dynamic sling until his residual knee contracture was fully corrected, after which a cylinder cast was applied, followed by a knee gaiter for long-term use. Mobilisation with a walking frame was gradual due to residual knee pain and weakness.
Radiographs revealed generalised skeletal lucency with absence of the carpal bones in the right wrist and destruction of the right elbow joint
We assessed his metabolic markers in view of his profound osteopaenia. Dual-energy X-ray absorptiometry showed a Z-score of -3.7sd, and he was subsequently treated with intravenous infusion of a bisphosphonate, zoledronic acid. Radiographs of the distal femur now show 'zebra lines', which have previously been described in association with pamidronate therapy8(Figure 7).

Discussion
The genetic osteolyses are a group of rare disorders which are characterised by radiolucency and progressive disappearance of components of the skeleton. The various forms of osteolysis have been defined and named in terms of the anatomical distribution of the abnormalities, the occurrence of extra-skeletal manifestations and the mode of genetic transmission. There is considerable phenotypic overlap and the syndromic boundaries are blurred. Nosological confusion has been compounded by the inconsistent use of eponymic designations for some forms of osteolysis. Advances in molecular technology have clarified the situation to some extent and it has become apparent that several apparently disparate entities result from different mutations within the same gene. This observation has led to the concept of a 'spectrum' of overlapping conditions which share a similar biomolecular pathogenesis.
The Torg-Winchester syndrome is a rare autosomal recessive form of osteolysis in which mutations in the matrix met-alloproteinase-2 gene (MMP2) at the chromosomal locus 16q13 are causative.5,6,7,9 The matrix metalloproteinases are a group of enzymes involved in collagen homeostasis. The exact mechanism of how this may lead to the osteolysis syndromes is unknown, but it is believed that abnormal collagen breakdown leads to abnormal osteoblast activity and subsequent osteolysis.5 In this condition the clinical and radi-ographic features range from osteolysis which predominates in the wrists and ankles (Torg form), to severe generalised osteolysis with skin nodules and a coarse facies (Winchester syndrome).2,4 A third condition comprising subcutaneous nodules, arthropathy and osteolysis (NAO syndrome) which has been documented in consanguineous families in Saudi-Arabia is now included in this spectrum.1,10 Despite a wide geographical distribution of case reports, the genetic osteolyses are extremely rare. Indeed, in more than 40 years of focus on skeletal disorders in South Africa, only three previous cases have been identified in this country.3
The affected boy whom we have studied has progressive osteolysis with phenotypic manifestations which are all components of the Torg-Winchester spectrum. In particular, predominant but asymmetrical osteolysis of the carpus with widening of the metacarpals, metatarsals and phalanges is suggestive of the former Torg syndrome. The involvement of the elbows and other skeletal regions, together with the joint contractures and muscle wasting are also variable components of this syndrome. Similarly, the severity and distribution of the osteolysis is in keeping with the former Winchester syndrome. Finally, the clavicular widening is characteristic of the NAO syndrome.10 The fact that the patient has features of all three forms of osteolysis, which were previously regarded as autonomous entities, is entirely in keeping with the concept of a phenotypic spectrum.1
Differential diagnosis
The most frequent disorders which enter into the differential diagnosis in South Africa include:
• Osteogenesis imperfecta type III
• Bruck syndrome
• Gorham osteolysis
• Polyarticular juvenile idiopathic arthritis
• Juvenile idiopathic osteoporosis
The main condition which enters into the differential diagnosis of the Torg-Winchester NAO spectrum of heritable osteolysis is osteogenesis imperfecta (OI). Diagnostic differentiation of these disorders is especially important in South Africa in view of the relatively high frequency of OI type III in the indigenous African population.11 The manifestations of this autosomal recessive disorder are skeletal fragility, which leads to multiple fractures, together with limb and spinal deformity and marked physical handicap. An unusual complication encountered in a few individuals with this form of OI is the occasional occurrence of flexion contractures of the limbs.
The pathogenesis of this combination, which is termed Bruck syndrome, has not yet been elucidated.12,13 The diagnosis of the osteolysis spectrum in the affected boy is strongly supported by the disappearance of the carpal bones, the configuration of the tubular bones of the hands and feet, and the occurrence of significant osteopaenia. An additional radiological discriminant includes the asymmetrical lytic destruction of the affected joints.9 In the affected boy, the radiological appearances were entirely in keeping with those of the Torg-Winchester osteolysis as depicted in the definitive Atlas of Genetic Disorders of Skeletal Development14 and the diagnosis was confirmed by an international expert in the radiology of the osteolyses.15
Management
There is a paucity of literature concerning the management of the orthopaedic manifestations of the genetic oste-olyses. The available literature suggests that the skeletal lysis is progressive, and it is appropriate that the management should focus on correction of deformities and prevention of recurrence.3 Social support by family members is essential to promote rehabilitation.
In the upper limb, the aim of treatment should be to obtain a functional limb to promote independence. Functional assessment by occupational therapists is invaluable in this respect. With regard to the elbow, management will differ depending on the presence of unilateral or bilateral deformities - unilateral deformities should allow functional use of the involved limb, e.g. to bring the hand to the mouth in order to eat; bilateral deformities should be corrected in order to allow eating and toileting tasks. This can be accomplished by serial casting. With regard to the wrist and hand, the focus is again on functionality - the wrist must attain a neutral position to perform activities of daily living. This can be attained by osteotomy and external fixation with gradual correction, or by serial casting. Phalangeal deformities should be managed with aggressive mobilisation and splinting.
In the lower limb, the focus should be on correction of deformities to allow independent mobilisation. In the event of knee flexion contractures, the correction of the deformity can be obtained using serial casting or reversed dynamic sling traction, an entity that was previously described to correct flexion contractures in haemophiliacs.16 The ankle if involved must remain plantigrade, as an equinus deformity will prevent mobilisation.
Bisphosphonate therapy
Bisphosphonates have been used in adults with osteoporosis in order to increase bone mass and decrease fracture risk in a broad spectrum of disease. In children, the indications for use in primary and secondary osteoporosis are not as well defined. There appears to be no consensus over the ideal agent, dosage or treatment duration for osteoporosis in children. In this child, initiation of bisphosphonate therapy was based on a low bone mineral density, and was in the form of a zoledronic acid infusion.17 However, pamidronate has been shown to be ineffective in improving osteolysis or osteoporosis in the appendicular skeleton of individuals with Torg-Winchester syndrome.18
Radiographs of this child's distal femur exhibit 'zebra lines' which were initially described in association with children on pamidronate therapy. These transverse stria-tions are typical of children receiving cyclical bisphospho-nate therapy, and reflect 'increased bone mineralisation, as an indirect response to the inhibition of osteoclastic activity at the time of drug treatment'.7 These have been termed 'zebra lines' in order to differentiate them from Harris growth arrest lines, which occur in the metaphyses of rapidly growing long bones following systemic disorders, e.g. malnutrition, femur fractures, infection.8 Femoral radiographs also indicate a prior Salter Harris II distal femur fracture, which followed insignificant trauma. This is in keeping with the underlying osteoporotic bone.
Comment
The Torg-Winchester form of osteolysis warrants consideration as a possible diagnosis in children presenting with clinical deformity following minimal trauma and radiological evidence of progressive osteolysis. The orthopaedic management is demanding and includes long-term bracing and night splinting in functional positions to avoid relapses.
PB received financial support from the National Research Foundation and the Medical Research Council of South Africa.
The content of this article is the sole work of the authors. No benefits of any form have been or are to be received from a commercial party related directly or indirectly to the subject of this article. Informed consent was obtained.
Ethical approval for investigation of the heritable connective tissue disorders in South Africa was received by PB from the UCT Health Sciences Faculty Research Ethics Committee ref 026/2010.
References
1. Warman ML,Cormier-Daire V, Hall C, Krakow D, Lachman R, Le Merer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, Robertson S, Savarirayan R, Sillence D, Spranger J, Unger S, Zabel B, Superti-Furga A. Nosology and Classification of Genetic Skeletal Disorders: 2010 Revision. Am J Med Genet Part A 2011;155:943-68. [ Links ]
2. Matthieson C, Faurholt Pedersen V, Helin P, Krag Jacobsen G, Soe Nielsen N. Winchester syndrome. Int Orthop 2001;25:331-33. [ Links ]
3. Beighton P, Mennen U, Golele SS, Urban MF. Orthopaedic implications of heritable osteolysis in South Africa. SA Orth Journal 2007;6(2):26-32. [ Links ]
4. Vanatka R, Rouzier C, Lambert JC, Leroux C, Coussement A. Winchester syndrome: the progression of radiological findings over a 23-year period. Skel Radiology 2011;40:347-51. [ Links ]
5. Zankl A, Bonafe A, Calceferra V, Di Rocco M, Superti-Furga A. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin Genet 2005;67:261-66. [ Links ]
6. Rouzier C, Vanatka R, Bannwarth S, Philip N, Coussement A, Pasquis-Flucklinger V, Lambert J-C. A novel homozygous MMP2 mutationin a family with Winchester syndrome. Clin Genet 2006;69:271-76. [ Links ]
7. Zankl A, Pachman L, Poznanski A, Bonafe L, Wang F, Shusterman Y, Fishman DA, Superti-Furga A. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester Syndrome. J Bone and Mineral Research 2007;22(2):329-33. [ Links ]
8. Al Muderis M, Azzopardi T, Cundy P. Zebra lines of pamidronate therapy in children. J Bone Joint Surg Am 2007;89:1511-16. [ Links ]
9. Gok F, Crettol LM, Alanay Y, Hacihadioglu B, Kocaoglu, Bonafe L, Ozen S. Clinical and radiographic findings in two brothers affected with a novel mutation in matrix metalloproteinase 2 gene. Eur J Pediatr 2010;169:363-67. [ Links ]
10. Al-Otaibi L, Al-Mayouf SM, Majeed M, al-Eid W, Bahabri S, Hugosson CO. Radiological findings in the NAO syndrome. Pediatr Radiol 2002;32:523-28. [ Links ]
11. Beighton P, Versfeld GA. On the paradoxically high relative prevalence of osteogenesis imperfecta type III in the Black populatioin of South Africa. Clin Genet 1985;27:398-401. [ Links ]
12. Viljoen D, Versfeld G, Beighton P. Osteogenesis imperfecta with congenital joint contracture (Bruck Syndrome). Clin Genet 1989;36:122-26. [ Links ]
13. Mokete L, Robertson A, Beighton P. Bruck syndrome: congenital joint con-tractures with bone fragility. J Orthop Sci 2005;10:641-46. [ Links ]
14. Spranger JW, Brill PW, Nishimura G, Superti-Furga A, Unger S. Bone dys-plasias. An Atlas of Genetic Disorders of Skeletal Development, 3rd edition. Oxford University Press, Oxford;2012:644-46. [ Links ]
15. Spranger JW. Personal communication. [ Links ]
16. Stein H, Dickson RA. Reversed Dynamic Slings for Knee-Flexion Contractures in the Hemophiliac. J Bone Joint Surg Am.1975;57(2):282-83. [ Links ]
17. Bachrach LK, Ward LM. Clinical review: Bisphosphonate use in childhood osteoporosis. J Clin Endocrinol Metab 2009:94(2):400-409. [ Links ]
18. Phadke SR, Ramirez M, DiFeo A, Martignetti JA, Girisha KM. Torg-Winchester syndrome: lack of efficacy of pamidronate therapy. Clin Dysmorphol 2007;16(2):95-100. [ Links ]
 Reprint requests:
Reprint requests:
Reprint requests:
Dr J Bertie
Department of Orthopaedics, Greys Hospital
Pietermaritzburg Private Bag 9001, Pietermaritzburg, 3200
Tel: (033) 897-3000 Fax: 086 5784 100 E
mail: juliabertie@yahoo.com


{kind=link}