










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSA Orthopaedic Journal
On-line version ISSN 2309-8309
Print version ISSN 1681-150X
SA orthop. j. vol.10 n.2 Centurion Jan. 2011
CASE REPORT AND REVIEW OF THE LITERATURE
Cleidocranial dysplasia presenting as familial coxa vara in a South African family
JD BertieI; D ThompsonII; P BeightonIII
IOrthopaedic Registrar, Greys Hospital, Pietermaritzburg
IIPrincipal Specialist, Paediatric Orthopaedics, Greys Hospital, Pietermaritzburg
IIIEmeritus Professor, Division of Human Genetics, Faculty of Health Sciences, University of Cape Town
ABSTRACT
BACKGROUND: We have studied a South African family in which four persons in three generations had bilateral coxa vara. The proband, a boy aged 4 years, presented with a disturbance of gait. His affected father and uncle had been assessed and operated on by the first named consultant (DT), and his grandmother by DT's father.
METHOD: We performed a general and orthopaedic examination as well as a radiologic skeletal survey of the above four family members.
RESULTS: The proband's clinical and radiological assessment showed facial features and clavicular changes in keeping with cleidocranial dysplasia, an autonomous autosomal dominant genetic disorder. Radiographs of his hips revealed bilateral coxa vara deformities. The proband's father, uncle and grandmother also had similar facial and clavicular features of cleidocranial dysplasia. Their hip radiographs showed evidence of previous internal fixation subsequent to proximal femoral osteotomies.
CONCLUSION: Cleidocranial dysplasia is relatively common in South Africa and this condition warrants consideration as a diagnosis in any person with familial coxa vara.
Key words: cleidocranial dysplasia, coxa vara, familial
Introduction
Cleidocranial dysplasia (CCD) [MIM 119600] is an autosomal dominant skeletal dysplasia which is characterised by defective formation of the clavicles and patency of the anterior fontanelles.1 Affected individuals have typical facial features, short stature and a varying spectrum of additional dental and minor skeletal abnormalities. Apart from undue mobility of the shoulder girdles, persons with CCD are often asymptomatic. Orthopaedic problems are infrequent, but may include pes planus, genu valgum, scoliosis and bilateral coxa vara.2
We have assessed four family members in three generations of a South African family who initially presented with bilateral coxa vara deformities. This abnormality had previously been successfully corrected surgically in three of these individuals. They also had clavicular and skull anomalies and a diagnosis of CCD was subsequently established following radiological studies. Two other persons in the kindred had the typical clavicular and cranial manifestations of CCD, but without coxa vara.
A large extended Cape Town family comprises several hundred persons with CCD.3 It is evident that this condition is relatively common in South Africa, and in the local context, CCD warrants consideration in the differential diagnosis of familial coxa vara. Our observations are presented, depicted and discussed in this article and the orthopaedic manifestations of CCD are reviewed.
Case report
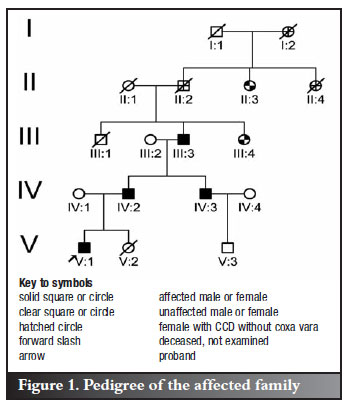
In the affected family, a boy, his father, paternal uncle and paternal grandmother each presented with bilateral coxa vara. In this setting of familial coxa vara (FCV), the diagnosis of CCD was subsequently established. Two females in generations II and III had CCD without FCV. A male cousin of the proband was unaffected. Other relatives were deceased and their diagnostic status remains unknown. The pedigree of the affected family is shown in Figure 1.
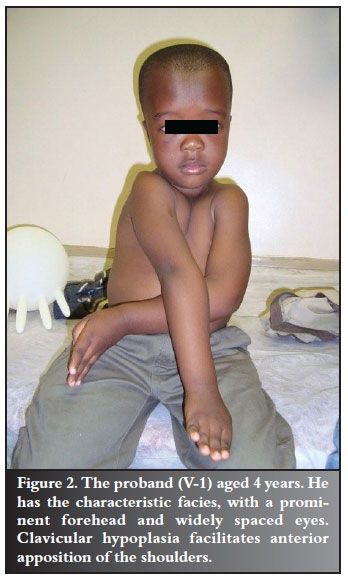
The proband (V-1) was a 4-year-old healthy boy of short stature, with growth-for-age measured along the tenth centile for height and the twenty-fifth centile for weight. He was initially seen with the complaint of not being able to run as fast as his peers. He had a patent anterior fontanelle, prominent forehead, flattened nasal bridge and eyes set widely apart. The sternal aspect of both clavicles was absent, and he was able to almost approximate his shoulders under his chin (Figure 2). He had a normal range of hip movements and no obvious spinal deformities.
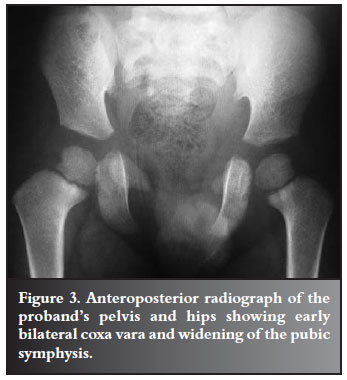
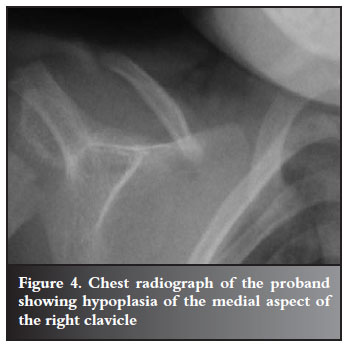
Radiological assessment of his pelvis showed early bilateral coxa vara with a femoral neck-shaft angle of 120º, and a Hilgenreiner epiphyseal angle greater than 45º. The pubic symphysis was widened (Figure 3) and the chest radiograph revealed bilateral partial absence of the medial aspects of both clavicles (Figure 4). Wormian bones were evident on the skull radiographs. The diagnosis of CCD was made on a basis of these clavicular and cranial abnormalities.
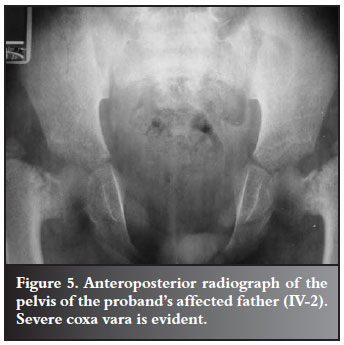
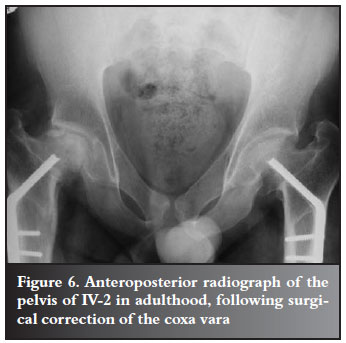
The proband's father (IV-2) and uncle (IV-3) had short stature together with the typical clavicular and cranial abnormalities of CCD. They both presented with bilateral coxa vara, for which osteotomies were performed by DT in 1985, at the ages of nine and six years respectively (Figures 5 and 6) and they are currently awaiting surgery for trochanteric advancement.
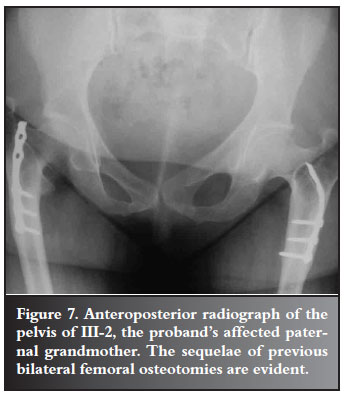
The proband's paternal grandmother (III-3) has short stature, with features in keeping with CCD, including an absent right clavicle and wormian bones in the cranium. Approximately 40 years ago, bilateral femoral osteotomies were undertaken by DT's father, and she is now largely asymptomatic (Figure 7). No further surgery is currently being contemplated.
Discussion
Affected individuals usually have mildly short stature; their length at birth is usually normal, but decreases to around the second centile by the age of 4-8 years. The limbs are disproportionately short compared to the trunks, with upper limbs affected more than lower limbs. General health is good, with normal mental capabilities and life expectancy.1-3
Cleidocranial dysplasia is an autosomal dominant condition and the determinant gene is localised at chromosome 6p21.2,4 The gene product is the transcription factor RUNX2 (CBFA1) which is important for both membranous and enchondral ossification.2 The defect in membranous ossification is reflected by the clinical involvement of mostly midline structures notably cranium, spine and pubic symphysis.2,4-6
Cranial and facial features of cleidocranial dysplasia
Typical facial features3
• large head with a small face
• bossing of frontal and parietal regions
• patent sutures and fontanelles, with delayed fontanelle closure
• increased distance between the eyes
• widened, flattened nasal bridge
• mandible prognathism
• dental complications, notably supernumary teeth, retained deciduous teeth and delayed eruption of permanent teeth.
Dental problems and these craniofacial characteristics are the main reasons for affected individuals seeking medical attention. For this reason, dentists and odontologists are often instrumental in making a positive diagnosis.5 Typical radiological features of the skull:1-5,7
• incomplete ossification of cranial sutures
• patent fontanelles
• wormian bones, especially in the parietal and occipital area
• underdeveloped maxilla and sinuses
Skeletal manifestations of cleidocranial dysplasia
Clavicle1-3,5-7
The clavicles may be entirely or partially absent, usually bilaterally, allowing shoulder hypermobility and subsequently leading to the classic sign of the individual being able to approximate the shoulders under the chin. These changes are very variable and in some affected persons, the clavicles may be normal. Clavicular abnormalities are seldom a cause of disability, except if compressing the brachial plexus.
The spectrum of clavicular abnormalities includes absent or hypoplastic lateral third (common), each clavicle consisting of two separate fragments, with the middle section being absent, absence of medial third of clavicle and pseudoarthrosis of the clavicle.
Congenital pseudoarthrosis of the clavicle is rare, but should be considered if there is unilateral involvement of especially the middle third of the right clavicle, and is present at birth. As a single entity, it is not associated with other musculoskeletal abnormalities.6 Pseudoarthrosis of the clavicle also warrants consideration when there is bilateral clavicular involvement in CCD. Right-sided pseudoarthrosis in CCD may occur in association with elevation of the first rib. As the development of the clavicular component of CCD is not adequately explained by inadequate membranous ossification, this anomaly may develop due to pressure exerted on the clavicle by the subclavian artery which is at a higher level on the right side. The presence of cervical ribs or elevated first ribs, both previously described in association with CCD, may contribute further to its development.6
Pelvis3,7
Pelvic anomalies include failure or delayed ossification of the pubic symphysis, failure of ossification of the ilium, a widened pubic symphysis and complete or incomplete absence of the sacrum or coccyx.
Hips3,7,8
Hip anomalies include coxa vara which is usually bilateral. Coxa vara is defined as any reduction in the normal angle between the femoral neck and shaft. In the newborn, the neck-shaft angle is 155º, and decreases with age to an angle of 130º.7 A 'chef's hat' appearance of the femoral head is suggestive but not pathognomonic of CCD.8
Other skeletal manifestations2,3,7
In the shoulder the scapulae may be small and dysplastic with abnormal supraspinatus fossa and acromial facets.
In the arm and forearm, the medial humeral epicondyle may be dysplastic, with partial absence of the radius.
In the hands, short, tapered distal phalanges are frequent. Accessory epiphyses, especially involving second metacarpal and cone-shaped epiphyses may be radiologically evident. In the feet, epiphyses present at both ends of metatarsals are common.
The spine is not usually malaligned but scoliosis, kyphosis and lordosis can occur.
Radiological features include prominent cervical transverse processes, cervical ribs, failure of union of neural arches, spondylolysis, spondylolisthesis and hemivertebrae.
Management of coxa vara in cleidocranial dysplasia
Surgical management of coxa vara is the gold standard and conservative management is regarded as being ineffective. In particular, surgery is needed to promote ossification of the femoral neck and correct the deformity.7,9 Absolute surgical indications include neck-shaft angle less than 90º, Hilgenreiner's epiphyseal angle greater than 60º, and documented progression of worsening of varus angulation.7,9,10
Timing of surgery has not been unanimously determined,10 but recurrence has been documented in children undergoing surgery before the age of 6 years. The optimal age must be determined individually - the child should not be too young in order to avoid early closure of the capital femoral epiphysis and subsequent leg length discrepancy and greater trochanteric advancement. Equally, the child should also not be too old, so that sufficient time remains for improvement of growth before growth plate closure.
In terms of surgical procedures, many different types of osteotomies have been described.9,10 The most reliable and least demanding procedure has been shown to be an intertrochanteric valgus osteotomy with plate fixation, the extent of which depends on the extent of the child's initial deformity.
In our case study, the natural history of the treated disease is apparent, with elongation of the greater trochanters causing difficulty with gait and hip movement. Epiphysiodesis of the greater trochanter has only previously been described in association with mild cases of coxa vara, but we are considering early growth retardation of the trochanteric apophyses of the proband.7
Conclusion
We have identified six individuals in four generations of a family with CCD, four of whom have bilateral coxa vara. The recognition of the characteristic facial, cranial and clavicular abnormalities is essential in order to make a diagnosis. It is imperative to identify hip pathology early on, in this instance bilateral coxa vara, in order to avoid deformity and delay in surgical management.
Acknowledgements
We are grateful to Dr Karen Fieggen, Division of Human Genetics, University of Cape Town, for her assistance. PB received financial support from the National Research Foundation and the Medical Research Council of South Africa.
References
1. Cooper SC, Flaitz CM, Johnston DA, Lee B, Hecht JT. A natural history of cleidocranial dysplasia. Am J Med Genet 2001;104:1-6. [ Links ]
2. Mundlos S. Cleidocranial dysplasia: Clinical and molecular genetics. J Med Genet 1999;36:177-82. [ Links ]
3. Jackson WP. Osteo-dental dysplasia (cleidocranial dysostosis); the 'Arnold head'. Acta Med Scand 1951;139:292-307. [ Links ]
4. Ramesar RS, Greenberg J, Martin R, Goliath R, Bardien S, Mundlos S, Beighton P. Mapping of the gene for cleidocranial dysplasia in the historical Cape Town (Arnold) kindred and evidence for locus homogeneity. J Med Genetics 1996;33:511-14. [ Links ]
5. Daskalogiannakis J, Piedade L, Lindholm TC, Sandor GKB, Carmichael RP. Cleidocranial Dysplasia: 2 Generations of Management. J Can Dent Assoc 2006; 72(4):337-42. [ Links ]
6. Lloyd-Roberts GC, Apley AG, Owen R. Reflections upon the etiology of congenital pseudoarthrosis of the clavicle with a note on cranio-cleido dysostosis. J Bone Joint Surg 1975;57-B:24-29. [ Links ]
7. Sharrard WJW. Paediatric Orthopaedics and Fractures. General Abnormalities of Skeletal Development. Blackwell Scientific Publications;1979:166-67. [ Links ]
8. Aktas S, Wheeler D, Sussman MD. The 'chef's hat' appearance of the femoral head in cleidocranial dysplasia. J Bone Joint Surg 2000;82B:404-408. [ Links ]
9. Babb FS, Ghormley RK, Chatterton CC. Congenital coxa vara. J Bone Joint Surg 1949;31-A:115-31. [ Links ]
10. Richie MF, Johnston II CE. Management of developmental coxa vara in cleidocranial dysostosis. J Paediatr Orthop 1989;12(7):1001-1004. [ Links ]
 Reprint requests:
Reprint requests:
Dr J Bertie
Department of Orthopaedics, Greys Hospital, Pietermaritzburg
Private Bag 9001 Pietermaritzburg 3200
Tel: (033) 897-3000 Fax: 086 5784 100
Email: juliabertie@yahoo.com
The content of this article is the sole work of the authors.
No benefits of any form have been or are to be received from a commercial party related directly or indirectly to the subject of this article.
Informed consent was received from the affected family.
Ethical approval for investigation of the heritable connective tissue disorders in South Africa was received by PB from the UCT Health Sciences Faculty Research Ethics Committee ref 026/2010.
The research has been approved by an ethical committee.

