










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSA Orthopaedic Journal
versión On-line ISSN 2309-8309
versión impresa ISSN 1681-150X
SA orthop. j. vol.9 no.1 Centurion ene. 2010
CLINICAL ARTICLE
Chondromyxoid fibroma - A case series and radiological review
J van HeerdenI; JC VerwayenII; DA BamIII
IMBChB(Pret),Registrar, Department of Radiology, Steve Biko Academic Hospital, University of Pretoria, Faculty of Health Sciences
IIMBChB(Pret), DMRD RCP(London), RCS(Eng), MMed RAD(Pret), Senior Consultant, Department of Radiology, Steve Biko Academic Hospital, University of Pretoria, Faculty of Health Sciences
IIIMBChB (Pret), Registrar, Department of Radiology, Steve Biko Academic Hospital, University of Pretoria, Faculty of Health Sciences
ABSTRACT
Chondromyxoid fibromas are rare, benign tumours that resemble cartilage, initially arising in the cortex of affected bones (most commonly the lower limbs).1,2
Their documented incidence is less than 1% of all primary bone tumours (approximately 2% of all benign bone tumours) with males and females being equally affected.1,2
This case series and radiological review demonstrates some of the typical findings associated with this tumour.
Introduction
Chondromyxoid fibromas are rare, benign tumours that consist of immature myxoid mesenchymal tissue with features of primitive cartilaginous differentiation.1,2
Patients most commonly affected tend to be in their second or third decades of life.1,2
The case series that follows depicts some of the classical findings associated with chondromyxoid fibromas.
Case series
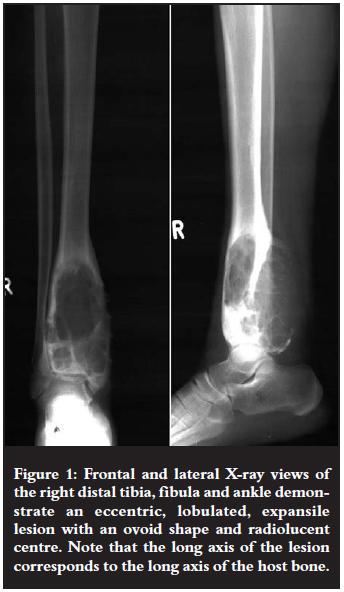
The 16-year-old female patient, shown in Figure 1, presented with a painful mass in the region of her right distal tibia.
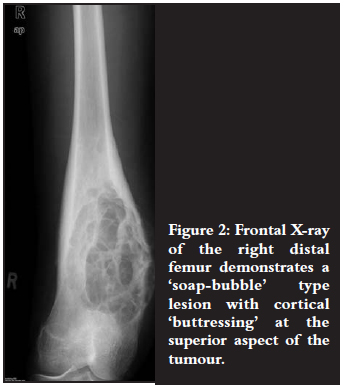
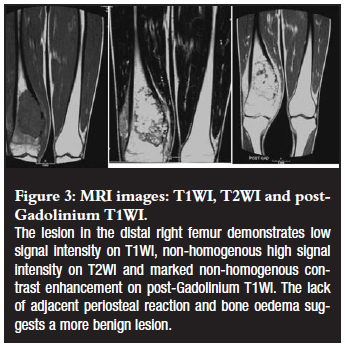
An 18-year-old male patient presented with a painful mass in the region of his right distal femur (Figures 2 and 3).
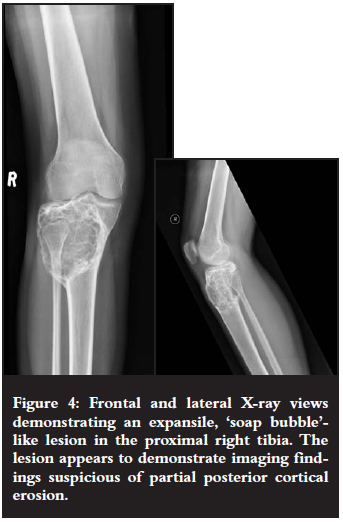
A 20-year-old female patient (Figure 4) presented with a mass in the region of her right proximal tibia.
Discussion
Chondromyxoid fibromas are rare, benign cartilage-like tumours. The initial clinical presentation varies from a mass (painful or painless, depending on the size) to a pathological fracture or as an asymptomatic incidental finding.
They occur predominantly in adolescent patients and young adults (patients in their second and third decades).2,3
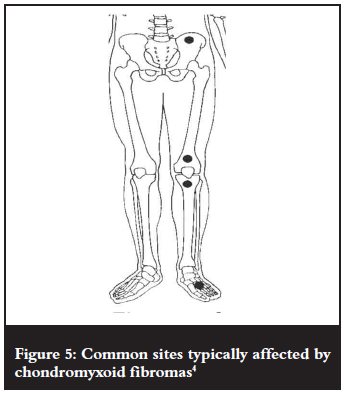
Common sites of occurrence (Figure 5) are the long bones of the lower limb (60%) especially the distal femur, proximal tibia and fibula.4 Other (less common) sites that can be affected include the short tubular bones of the hands and feet (20%) and the flat bones such as the pelvis and ribs (<20%).2,3 They present as eccentric lesions that may be metaphyseal (47%), meta-diaphyseal (20%), meta-epiphyseal (26%), diaphyseal (4%) or epiphyseal (3%), resulting in thinning and expansion of the adjacent bony cortex.3
The surgical staging for benign musculoskeletal tumours can also be used when evaluating these tumours:2
• Stage 1: Latent
• Stage 2: Active
• Stage 3: Aggressive
On X-ray views they appear as eccentric and lobulated (soap bubble-like), expansile lesions.1-4 These tumours are characteristically ovoid in shape with sclerotic margins and radiolucent centres.3 Partial cortical erosion may be present.3 The long axis of the lesion is usually parallel to the long axis of the host bone.3,4 Septations within the lesion may mimic trabeculations.3 These tumours do not commonly demonstrate matrix calcifications or soft tissue extension/swelling.2-4 Cortical 'buttressing' at the junction where the expanding tumour abuts against the cortex may be seen.1
Computed tomography (CT) can be used to further study the nature and extent of the tumour. It is the best imaging modality for detecting sclerotic tumour margins and ridges as well as matrix mineralisation.2,4 CT also readily depicts the cortical integrity of the lesion. Chondromyxoid fibromas demonstrate enhancement on post-contrast CT views.2,4
Magnetic resonance imaging (MRI) is the preferred imaging modality for the evaluation of the true soft tissue extent of the lesion in order to aid with pre-operative planning and postoperative confirmation of complete resection.1,4 Chondromyxoid fibromas usually demonstrate low signal intensity on T1WI, heterogeneous high signal intensity on T2WI (most likely due to varying amounts of chondroid, myxoid and fibrous tissue) and high signal intensity on STIR (fat suppression) sequences.2,4
Non-homogenous enhancement following intravenous administration of Gadolinium DTPA is typically noted (due to mixed tissue nature described above).2,4
On Technetium-labelled bone scan views, the tumour demonstrates increased radiotracer uptake.2 Should angiography be performed, the tumour characteristically demonstrates minimal angiogenesis.2
The treatment in most of the reported cases consists of intralesional curettage.4 Curettage with bone graft or osseous cement on its own has a high incidence of recurrence, up to 25%.6 Fotiadis et al. reported that curettage with a curette and a high frequency burr as well as osseous cement with a Kirschner needle leads to an improvement of bone stamina and a decreased rate of recurrence.7
Malignant degeneration is distinctly unusual. However, there have been isolated case reports describing malignant degeneration following radiotherapy.3,5 Thus, irradiation as a mode of therapy is contraindicated.5
The differential diagnosis includes:
• simple bone cyst1,3,4
• aneurysmal bone cyst - usually demonstrates fluid-fluid levels and periosteal new bone formation without matrix mineralisation2
• Non-ossifying fibroma - usually no cortical ballooning or cortical erosion2
• Fibrous dysplasia - usually at a central location without internal septations. The peak incidence of osteofibrous dysplasia occurs in the first decade of life and the lesions typically demonstrate more sclerosis2
• Giant cell tumour - expansile, lytic tumour that usually extends to the subchondral bone2
• Enchondroma - more classically involves the hands and feet4
• Chondroblastoma - usually epiphyseal lesions with calcified matrix in approximately 50 per cent of tumours2
Conclusion
Chondromyxoid fibromas are rare tumours that should form part of the differential diagnosis to be considered in expansile, ovoid bone tumours with a 'soap bubble'-like appearance and cortical buttressing along the tumour margins.
The final, definitive diagnosis is made by clinical, radiological and pathological evaluation of tumour characteristics. Histologically the tumour consists of primitive cartilaginous tissue, fibrous tissue as well as immature myxoid tissue that may, histologically, mimic a chondrosarcoma - thus necessitating imaging modalities (X-ray, CT and MRI) to aid in the final diagnosis.2
In the absence of exposure to radiotherapy, the potential of malignant degeneration of chondromyxoid fibromas is rare.
Acknowledgements
Prof ZI Lockhat, University of Pretoria - Faculty of Health Sciences, Head: Department of Radiology, Steve Biko Academic Hospital.
References
1. Adam A, Dixon AK. Grainger & Allison's Diagnostic Radiology. 5th ed. In: Stoker DJ, Saiffudin A. Bone Tumors: General Characteristics and Benign Lesions. Philadelphia: Elsevier Churchill Livingstone; 2008: 1039-41. [ Links ]
2. Stoller DW, Tirman PFJ, Bredella MA. Diagnostic Imaging Orthopaedics. Salt Lake City: Amirsys; 2004: 8/30-8/33. [ Links ]
3. Dähnert W. Radiology Review Manual. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2007: 57-8. [ Links ]
4. Greenspan A. Orthopedic Imaging. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2004: 617-22. [ Links ]
5. Scott S, George J. Chondromyxoid Fibroma. [Data base on Internet]. eMedicine. [Updated: 2009/03/29; Sited: 2009/04/30]. Available from: http://emedicine.medscape.com/article/388738. [ Links ]
6. Durr HR, Lienemann A, et al. Chondromyxoid fibroma of bone. Arch Orthop Trauma Surg (2000)120:42-47. [ Links ]
7. Fotiadis E, Akritopoulus P, et al. Chondromyxoid fibroma: a rare tumour with an unusual location. Arch Orthop Trauma Surg (2008)128:371-375. [ Links ]
 Reprint requests:
Reprint requests:
Dr J van Heerden
Private Bag X169 Steve Biko Academic Hospital Department of Radiology Level 5
Bridge E, Room 51103 Pretoria 0001
E-mail: jolandivh@hotmail.com
Tel: (012) 354-2406 Fax: (012) 354-2771
No benefits of any form have been received from a commercial party related directly or indirectly to the subject of this article. The content of this article is the sole work of the authors.

