










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouthern African Journal of Critical Care (Online)
On-line version ISSN 2078-676X
Print version ISSN 1562-8264
South. Afr. j. crit. care (Online) vol.36 n.2 Pretoria Nov. 2020
http://dx.doi.org/10.7196/SAJCC.2020.v36i2.420
CASE SERIES
Haemophagocytic lymphohistiocytosis: Five years' experience at tertiary hospitals in Free State Province, South Africa
M NienkemperI; J MalherbeII; C BarrettIII
IBSc, MB ChB, MMed (Int Med), FCP; OrcID 0000-0002-7590-7841; Department of Internal Medicine, School of Clinical Medicine, Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
IIMB ChB, MMed (Int Med), Cert Clin Haem; OrcID 0000-0002-7184-5081; Department of Internal Medicine, School of Clinical Medicine, Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
IIIMB ChB, MMed (Int Med), FCP; OrcID 0000-0001-7700-8893; Department of Internal Medicine, School of Clinical Medicine, Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
ABSTRACT
BACKGROUND. Haemophagocytic lymphohistiocytosis (HLH) is a potentially life-threatening syndrome if not recognised and managed early. It involves an uncontrolled pathological activation of the immune system, and it is either genetic or acquired. It presents with clinical and laboratory features of severe inflammation. Early initiation of effective therapy may reduce mortality from 95% to 35%.
OBJECTIVE. To raise awareness of HLH among healthcare professionals, particularly intensivists.
METHODS. We report nine cases of secondary HLH seen at tertiary hospitals in Bloemfontein, South Africa.
RESULTS. All patients presented with fever, hypertriglyceridaemia, hyperferritinaemia, transaminitis and cytopenia. Haemophagocytosis was noted on bone marrow biopsy in 66.7% (n=6/9) of the patients. More than one-third (44.4%; n=4/9) of the cases were triggered by a lymphoma, 44% (n=4/9) were associated with infection and 11% (n=1/9) were associated HIV. Finally, 11.1% (n=1) of the patients were triggered by an underlying autoimmune disease. More than half (55.6%; n=5/9) of the cases had a fatal outcome.
CONCLUSION. A high index of suspicion may promote the accurate diagnosis of HLH in patients presenting with fever, transaminitis and unexplained cytopenia.
Keywords: haemophagocytic lymphohistiocytosis; haemophagocytosis; hyperferritinaemia; cytopenia; transaminitis.
The first case of haemophagocytic lymphohistiocytosis (HLH) was described in 1939 by Scott and Robb-Smith.[1] After their patients had initially been diagnosed with 'atypical Hodgkin disease', they named the condition histiocytic medullary reticulosis.[1,2] It was later classified as a type of malignant histiocytosis.^ Altook et al.[4] described HLH as 'a rare disease of massive, dysregulated cytokine release and secondary multi-organ failure [...] associated with high mortality'.[4]
HLH is classified according to aetiology, namely primary (genetic) or secondary (acquired).[5] Patients with primary HLH have an underlying genetic cause or a history of familial predisposition. These underlying causes involve genetic mutations that eradicate proteins crucially required for normal function of cytotoxic T-cells and natural killer (NK) cells.[6] In 70 - 80% of cases, primary HLH occurs in infants and young children,[7] with symptoms presenting within the first year of life, and 10% of cases diagnosed in the neonatal period.[8-10] However, cases have been described in older children and adults.[11] Secondary HLH occurs in the absence of a known genetic lesion and is triggered by an underlying condition resulting from a malignant, infectious, or autoimmune stimulus.[6] The secondary causes of HLH are summarised in Table 1. However, it is increasingly recognised that some patients with secondary HLH may have an underlying genetic predisposition.[4,7,11]

The clinical features of HLH are characterised by the overstimulation of macrophages and CD8+ T lymphocytes. The cytokines that are produced by the macrophages lead to a persistent state of hypercytokinaemia and a subsequent systemic hyperinflammatory syndrome. These cells proliferate, migrate and infiltrate various organs, resulting in progressive organ dysfunction and ultimately death. The liver, nervous system and bone marrow are commonly involved.[12,13]
Apart from the fact that the diagnosis of HLH may be challenging,[14,15-these patients are often gravely ill with rapid deterioration of their condition,[14] requiring admission to intensive care units (ICUs). Patients presenting with a sudden onset of unexplained systemic inflammatory response syndrom, including features of malaise, jaundice, fever, generalised lymphadenopathy and hepatosplenomegaly, are highly suspicious of having HLH.[16] The Familial HLH Study Group of the Histiocyte Society developed the criteria for the diagnosis of HLH[11,17,18] that are shown in Table 2.

The presence of haemophagocytosis in bone marrow, spleen or lymph nodes is the key pathological finding in HLH. However, diagnosis does not depend on the presence of haemophagocytosis, as it has not been consistently present in patients with documented HLH.[11] Haemophagocytosis is often a cyclical process and it may be absent on the initial bone marrow investigation.[17]
HLH is associated with multi-organ involvement. As the liver is the most commonly affected organ, it is problematic to diagnose HLH in the absence of increased transaminases.[12] Other features include conjugated hyperbilirubinaemia, transaminitis leading to fulminant liver failure, hypoalbuminaemia, hyponatraemia, elevated D-dimers and lactate dehydrogenase, hypotension and renal or respiratory failure. Central nervous system involvement is also possible.[12]
In addition to the underlying cause, the treatment of HLH depends on the severity of the patient's condition.[19] Based on the HLH-94 protocol proposed by the Histiocyte Society in 1994,[20] induction therapy of 8 weeks consists of etoposide (VP-16) at a dose of 150 mg/m2 intravenously, given twice a week for the first 2 weeks and once a week for the remaining 6 weeks. Dexamethasone is administered intravenously or orally, starting at a dose of 10 mg/m2 daily and tapered over 8 weeks. For patients with central nervous system involvement, intrathecal methotrexate and hydrocortisone are also administered.[20,21]
Allogeneic stem cell transplantation is required in patients with central nervous system involvement, an HLH gene mutation and persistent or relapsing secondary disease. Patients destined for transplant are maintained on pulses of etoposide and dexamethasone combined with cyclosporine.[20,21] Stem cell transplant in patients with familial HLH improves the 3-year survival rate from 0% to 50%, and is potentially curative. Without treatment, the prognosis of genetic HLH is poor, with a median survival rate of only 1 - 2 months.[n
HLH that is triggered by the Epstein-Barr virus (EBV) has been found to have the worst outcomes, with mortality ranging from 25 - 100%.[22,23] Since the introduction of etoposide in the therapeutic regimen, good results have been observed, especially when initiated within the first 4 weeks of treatment. Active EBV infection with >10 000 copies μg of cellular EBV DNA can also be treated with weekly rituximab for 1 - 4 weeks. The duration of treatment depends on decreasing levels of EBV DNA.[11]
The primary aim of this report was to raise awareness of HLH among critical care physicians, since the early diagnosis and treatment of HLH may affect the outcome of this condition.
Methods
We describe 9 cases of acquired HLH diagnosed over a 5-year period (2012 - 2016) at 2 tertiary hospitals (Universitas Academic Hospital and Pelonomi Tertiary Hospital) in Bloemfontein, South Africa. We describe the patients' clinical presentation, aetiology and outcome. All clinical information was obtained from the hospitals' computerised database and patients' clinical files. Approval to report these cases was obtained from the Health Sciences Research Ethics Committee of the University of the Free State (ref. no. UFS-HSD2017/0309).
Description of cases
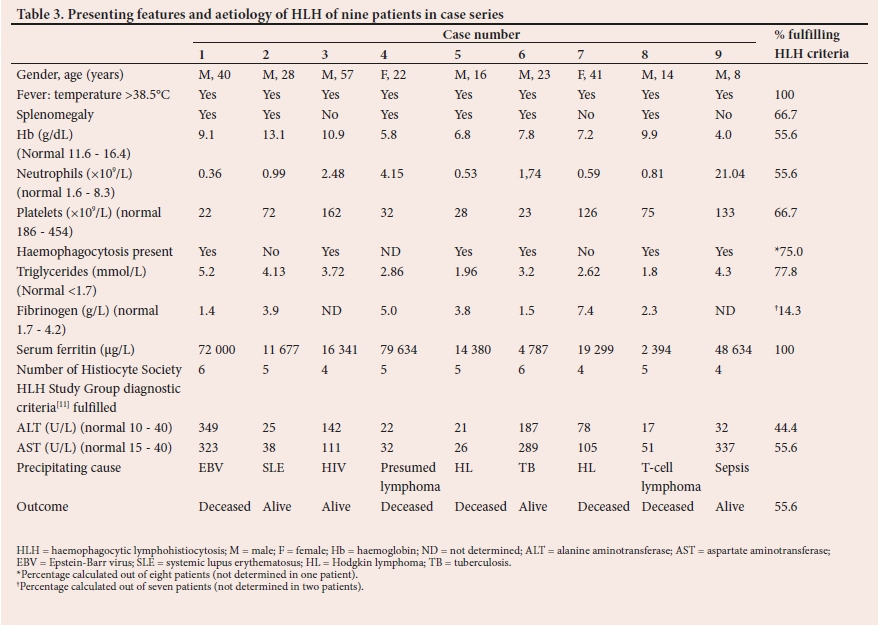
All cases fulfilled the criteria for the diagnosis of HLH according to the HLH Study Group diagnostic criteria.[11,17,18] The nine cases are summarised in Table 3. The majority of the cases (88.8%; n=8/9) were HIV-negative and 11.1% (n=1/9) were HIV-positive. A summary of the full blood count results of all patients is documented in Table 3.
Case 1
A previously well 40-year-old male presented with night sweats, fever, haemoptysis, abdominal distention and early satiety. The patient was pale, pyrexial, had scleral icterus, palpable hepatosplenomegaly and bilateral inguinal lymphadenopathy, and signs in keeping with a right lower lobe pulmonary consolidation were noted. The liver enzymes were suggestive of intrahepatic cholestasis. He was admitted and treated for community-acquired pneumonia (CAP), but showed no response to antibiotic therapy. His blood and urine cultures were negative. No sputum culture could be obtained from the patient.
Treatment according to the HLH-94 protocol was initiated within 3 days of admission. The patient showed a good clinical response with resolution of fever, but developed neutropaenic sepsis secondary to etoposide on day 28 after admission and died. An EBV viral load of 274 000 copies/mL was determined during postmortem, and confirmed that the viral infection was the trigger of HLH.
Case 2
A 28-year-old male presented with a 3-month history of night sweats, rigors and flu-like symptoms as well as painful joints experienced for 2 months. No previous medical history of significance was reported. The patient was pyrexial and had multiple enlarged anterior triangle cervical lymph nodes and a palpable splenomegaly. A lymph node biopsy showed reactive follicular hyperplasia without features of lymphoma. Bone marrow examination was negative for both Mycobacterium tuberculosis and malignancy. Due to clinical deterioration and fulfilment of five out of eight Histiocyte Society HLH Study Group diagnostic criteria, the HLH-94 treatment protocol was initiated.
During the work-up for precipitants of HLH, findings that confirmed previously undiagnosed systemic lupus erythematosus (SLE) were reported as the trigger of HLH. Induction therapy with etoposide and dexamethasone was administered for 8 weeks and yielded a positive response. He developed lupus nephritis 2 months later that was successfully treated with cyclophosphamide. HLH maintenance therapy with cyclosporine was not initiated, and he remained well on follow-up.
Case 3
A 57-year-old HIV-infected male patient with a CD4 count of 333 cells/μL and a suppressed HIV viral load presented with fever, abdominal pain, nausea and vomiting. His antiretroviral therapy was a combination tablet consisting of emtricitabine, tenofovir and efavirenz. The patient was pyrexial with rigors and had scleral icterus. A chest examination revealed scattered crepitations. He had no palpable lymphadenopathy or hepatosplenomegaly.
A presumptive diagnosis of pulmonary tuberculosis (TB) was made, and he was started on standard TB treatment as well as amoxicillin/ clavulanic acid for an underlying CAP. A sputum TB polymerase chain reaction test was negative. TB treatment was discontinued after 3 weeks owing to a poor clinical response and persistent fever. Other causes for the persistent fever were investigated and none were found. The patient fullfilled four of the Histiocyte Society HLH Study Group diagnositc criteria. Within 24 hours of treatment according to the HLH-94 protocol, the fever subsided and he was discharged after completing the 8-week induction course. The only identifiable trigger for HLH was the underlying HIV infection. He remained well after 3 years of follow-up.
Case 4
A 22-year-old female presented with symptomatic anaemia and a 3-month history of disabling dizziness, malaise, dyspnoea, coughing and night sweats. She had no previous medical history and was not using any medication. She appeared chronically ill, was febrile and pale with anasarca and palpable hepatosplenomegaly. Chest radiography revealed a widened mediastinum with bilateral pleural effusions.
Persistent fever was associated with rising inflammatory markers, and urine and blood cultures remained negative. A computed tomography (CT) scan revealed large supraclavicular and paratracheal lymph nodes, splenomegaly and bilateral pleural effusions, which was in keeping with the working diagnosis of lymphoma, Ann Arbor stage III. Her Glascow Coma Score (GCS) deteriorated to 12/15. A CT scan of the brain revealed a central pontine hypodensity that was attributed to demyelination. She was transferred to the ICU with shock and disseminated intravascular coagulation, where she died within a day despite immediate treatment with antibiotics, etoposide and dexamethasone. Due to her rapid death, no histological diagnosis could be made. The patient fullfilled five of the Histiocyte Society HLH Study Group diagnostic criteria.
Case 5
A 16-year-old male with a history of Hodgkin lymphoma (HL) presented with complaints of drenching night sweats, fever, weight loss, severe fatigue and a productive cough. He had completed standard ABVD (doxorubicin, bleomycin, vinblastine, dacabazine) chemotherapy 8 years prior to this admission. On examination, he was pyrexial with pallor and splenomegaly. A bone marrow aspiration and biopsy was performed, which confirmed relapsed HL. The patient fullfilled five of the Histiocyte Society HLH Study Group diagnostic criteria.
Etoposide and dexamethasone were initiated with a rapid decline in serum ferritin values and good clinical response. He developed neutropaenic septic shock and was transferred to the high care unit for 13 days, after which he continued treatment for HL. He died 1 week later and the cause of death was documented as transfusion-related acute lung injury.
Case 6
A 23-year-old male presented with pancytopaenia, transaminitis, fever, severe vomiting and coughing. Examination revealed pyrexia, tachycardia and left lower lobe pulmonary consolidation, and the tip of the spleen was palpable. He was treated for CAP. Hepatosplenomegaly without intra- or extrahepatic bile duct dilatation was found on ultrasonography. A bone marrow biopsy was performed to investigate the pancytopaenia and showed overt haemophagocytosis, which prompted further investigation for HLH. He fulfilled six of the eight criteria and was treated according to the HLH-94 protocol. At that time a definitive trigger for HLH was not identified.
A month later, he developed marked neck stiffness with confusion and a GCS of 13/15. Lumbar puncture results were suggestive of TB meningitis, and TB treatment was initiated. Cultures later confirmed the diagnosis of TB. He showed a full recovery and remained well until he was lost to follow-up at 20 months when he relocated abroad.
Case 7
A 41-year-old female was referred with a possible diagnosis of stage IV HL. She was pyrexial and pale with palpable hepatomegaly and purpura on both lower limbs. A bone marrow biopsy confirmed stage IV HL with prominent granulomatous transformation. She further developed pancytopaenia and transaminitis, and her initial serum ferritin value of 1 720 μg/L increased to 19 299 μg/L within 10 days.
She fulfilled four of the Histiocyte Society HLH Study Group diagnostic criteria. The patient was initiated on standard ABVD chemotherapy for HL, but due to further deterioration, HLH-directed therapy was added to her chemotherapy, after which she stabilised. The HLH-94 therapy was stopped after 4 weeks and she only continued with ABVD as an outpatient. Two months after discharge, she presented with clinical deterioration associated with renal failure. Due to a serum ferritin level of 3 673 μg/L, a relapse of HLH was suspected. She was admitted to the high care unit, where acute dialysis was initiated and etoposide restarted. Despite these interventions, she died 2 days after admission.
Case 8
A 14-year-old male presented with a history of headache, poor appetite and dizziness. He also had an episode of epistaxis and gingival bleeding. On examination, he had generalised pathological lymphadenopathy and fever. Lymph node biopsy was compatible with a viral infection and initial bone marrow did not reveal any malignancy. He was admitted and treated for neutropaenic sepsis. He had persistent fever spikes, increased serum ferritin and triglyceride levels, pancytopaenia and splenomegaly on CT scan. A diagnosis of HLH was made based on five of the Histiocyte Society HLH Study Group diagnostic criteria.
He responded well to therapy as per HLH-94 protocol, and was discharged for follow-up at the paediatric oncology clinic. At follow-up, he presented with renal failure and severe hypercalcaemia. He died in the ICU a day later due to complications of severe hypercalcaemia, renal failure and right heart failure. On postmortem, a diagnosis of T-cell lymphoma was made.
Case 9
An 8-year-old male presented with persistent fever, weakness and vomiting that was unresponsive to antibiotics. He had pancytopenia and transaminitis. Examination showed pallor, jaundice, signs of dehydration, petechiae on his upper limbs and generalised lymphadenopathy. He had a high triglyceride value of 4.3 mmol/L. A bone marrow biopsy showed pronounced haemophagocytosis. An Enterococcus species was cultured from his urine, which subsequently led to acute renal failure and septic shock. The patient was transferred to the ICU and started on inotropes and dialysis. Etoposide, dexamethasone and meropenem were initiated in the ICU.
He responded well to the 8-week course according to the HLH-94 protocol. He was lost to follow-up. However, the mother was contacted and she reported that the child was doing well.
Discussion
Malignancy was identified as a trigger for HLH in 44.4% (n=4/9) of the cases, and these were HL (n=2), T-cell lymphoma (n=1) and unconfirmed lymphoma (n=1). Three cases were triggered by infection, namely EBV (n=1), TB (n=1) and Gram-negative bacteraemia (n=1). One case was triggered by undiagnosed SLE. The HIV prevalence in this case series was in keeping with the national HIV prevalence of 12.6%.[24] Of note is that the HIV-infected patient in our study had a suppressed viral load, leading to uncertainty about the HIV association with HLH. Furthermore, no infectious cause could be identified for his persistent pyrexia. HLH in HIV-infected patients is commonly triggered by concomitant opportunistic infections.[25,26] An underlying genetic predisposition could not be ruled out.
The patient in case 1 had overwhelming EBV infection with a high viral load, which increases the possibility that he had underlying X-linked proliferative disease (XLP). EBV is the most common trigger of HLH in patients with XLP. Rare cases in adulthood have been described.[27] Regrettably, no postmortem samples could be obtained from this patient to test for XLP.
Soluble CD25 and NK cell activity is part of the HLH Study Group diagnostic criteria, as shown in Table 2. However, these investigations are not available in our setting and were therefore not included as part of our diagnostic work-up. Hejblum et al.[28]conducted a Delphi study among 24 experts from 13 countries as a step toward defining international guidelines for the diagnosis of secondary HLH in adults. This study confirmed that soluble CD25 and NK cell activity is not available to most clinicians and is therefore usually not helpful in the diagnosis of HLH in adults. The most useful features were cytopenias, haemophagocytosis on histology, hyperferritinaemia, fever, organomegaly, raised LDH and an underlying predisposing disease.[28]
Elevated serum ferritin occurred in all patients (Table 3) with a mean value of 29 861 μg/L. In adults, a markedly raised serum ferritin is less specific for HLH than in children. Other causes include rheumatological disease, liver disease and haemochromatosis.
It is apparent that fever and hyperferritinaemia occurred in all patients diagnosed with HLH in Bloemfontein. We compared our findings with a similar local and international case series reported in the literature (Table 4) and our findings were consistent with the diagnostic criteria of the HLH Study Group. In two case series reports from Johannesburg[5] and the USA,[13] 80% of patients met 5 or more of the diagnostic criteria, compared with 66.7% of the patients in our study that presented with 5 or more of the criteria. This might be due to the unavailability of testing for NK cell activity and measurement of soluble CD25 in our setting.
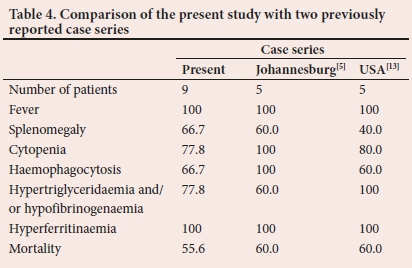
Transaminitis, although not part of the diagnostic criteria for HLH, was present in all nine patients, which was consistent with the case series from the USA.[29] Liver enzymes were not reported in the Johannesburg case series.[5] Liver dysfunction can occur as a result of infiltrating histiocytes and subsequent hepatocyte damage.[51 A diagnosis of HLH should be made with caution in a patient without transaminitis.[7]
Our mortality rate was similar to the other published HLH case series.[5,29] The high mortality rate could be ascribed to the fact that HLH mimics a variety of common medical conditions, leading to a delay in diagnosis, late presentation and delayed referral. Treatment-related mortality, mainly due to neutropenic sepsis, contributes to the poor outcomes in this condition.
In a meta-analysis of 35 case series that included 409 patients, the high mortality rate in patients with HLH was emphasised. Infection-related HLH has a poor outcome compared with autoimmune-related HLH. Favourable prognostic features include children and patients <50 years of age, no coagulopathy or disseminated intravascular coagulation, rapidly subsiding fever and early identification and initiation of treatment.[30]
Conclusion
The diagnosis of HLH remains challenging. Due to the nonspecific presentation of HLH, the differential diagnosis is often very broad. The survival of patients with HLH depends on early diagnosis and initiation of prompt and appropriate treatment. Adequate supportive care is imperative.
Since these cases were investigated in 2017, at least five other cases of HLH have been diagnosed at our centre. Physicians should consider HLH in the differential diagnosis of any patient presenting with fever and cytopenias. We suggest that a serum ferritin be obtained as part of the initial workup, and a diagnosis of HLH actively pursued if this value exceeds 1 000 μg/L. Investigations should also be directed at determining any underlying trigger. In critically ill or unstable patients, it may occasionally be prudent to initiate HLH directed therapy even while waiting for laboratory investigations.
Declaration. This study was performed in partial fulfilment of the MMed (Internal Medicine) degree of MN.
Acknowledgements. We would like to thank Prof. David Stones of the Department of Paediatric Oncology and Dr Cloete van Vuuren of the Department of Internal Medicine in the Faculty of Health Sciences at the University of the Free State for their contributions. We would also like to thank Dr Daleen Struwig from the Faculty of Health Sciences at the University of the Free State for technical and editorial presentation of the manuscript.
Author contributions. MN wrote the protocol, collected and analysed data and wrote the manuscript. JM and CB supervised this study, guided the process throughout and revised the manuscript. All authors approved the final version of this article for publication.
Funding. None.
Conflicts of interest. None.
References
1. Scott RB, Robb-Smith AHT. Histiocytic medullary reticulosis. Lancet 1939;234(6047):194-198. https://doi.org/10.1016/S0140-6736(00)61951-7 [ Links ]
2. Cetica V, Sieni E, Pende D, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry. J Allerg Clin Immunol 2016;137(1):188-196. [ Links ]
3. George MR. Hemophagocytic lymphohistiocytosis: Review of etiologies and management. J Blood Med 2014;5:69-86. https://doi.org/10.2147/JBM.S46255 [ Links ]
4. Altook R, Ruzieh M, Singh A, et al. Hemophagocytic lymphohistiocytosis in the elderly. Am J Med Sci 2019;357(1):64-74. https://doi.org/10.1016/j.amjms.2018.07.004 [ Links ]
5. Price B, Lines J, Lewis D, Holland H. Haemophagocytic lymphohistiocytosis: A fulminant syndrome associated with multiorgan failure and high mortality that frequently masquerades as sepsis and shock. S Afr Med J 2014;104(6):401-406. https://doi.org/10.7196/SAMJ.7810 [ Links ]
6. Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol Mech Dis 2018;13(1):27-49. https://doi.org/10.1146/annurev-pathol-020117-043625 [ Links ]
7. Jordan MB, Allen CE, Weitzman S, Filipovich A, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood 2011;118(15):4041-4052. https://doi.org/10.1182/blood-2011-03-278127 [ Links ]
8. Broglie L, Vitola B, Thakar MS, et al. Hemophagocytic lymphohistiocytosis mimicking neonatal hemochromatosis. Pediatr Hematol Oncol 2019:1-6. https://doi.org/10.1080/08880018.2019.1654051 [ Links ]
9. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med 2012;63(1):233-246. https://doi.org/10.1146/annurev-med-041610-134208 [ Links ]
10. McLean J. Katebian R, Suh E, Mirza K, Amin S. Neonatal hemophagocytic lymphohistiocytosis. Neoreviews 2019;20(6):e316-e325. https://doi.org/10.1542/neo.20-6-e316 [ Links ]
11. Tothova Z, Berliner N. Hemophagocytic syndrome and critical illness: New insights into diagnosis and management. J Intensive Care Med 2015;30(7):401-412. https://doi.org/10.1177/0885066613517076 [ Links ]
12. Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program 2011;1:178-183. https://doi.org/10.1182/asheducation-2011.1.178 [ Links ]
13. Thomas D, Shah N, Patel H, Pandya T, Gauchan D, Maroules M. Hemophagocytic lymphohistiocytosis: A series of five clinical cases in adult patients at a single institution with a review of the literature. N Am J Med Sci 2015;7(9):415-420. https://doi.org/10.4103/1947-2714.166225 [ Links ]
14. Tong QJ, Godole MM, Biniwale N, Jamshed S. An elusive diagnosis: Case reports of secondary hemophagocytic lymphohistiocytosis and review of current literature. Cureus 2019;11(4):e4548. https://doi.org/10.7759/cureus.4548 [ Links ]
15. Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NaCHO). Pedriatr Blood Cancer 2019:e27929. https://doi.org/10.1002/pbc.27929 [ Links ]
16. Rosado FGN, Kim AS. Hemophagocytic lymphohistiocytosis: An update on diagnosis and pathogenesis. Am J Clin Pathol 2013;139(6):713-727. https://doi.org/10.1309/AJCP4ZDKJ4ICOUAT [ Links ]
17. Henter JI, Samuelsson-Horne AC, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002;100(7):2367- 2373. https://doi.org/10.1182/blood-2002-01-0172 [ Links ]
18. Henter JI, Horne AC, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for heamophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124-131. https://doi.org/10.1002/pbc.21039 [ Links ]
19. Esteban YM, de Jong JL, Tesher MS. An overview of hemophagocytic lymphohistiocytosis. Pediatr Ann 2017;46(8):e309-e313. https://doi.org/10.3928/19382359-20170717-01 [ Links ]
20. Trottestam H, Horne A, Aricò M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: Long-term results of the HLH-94 protocol. Blood 2011;118(17):4577-4584. https://doi.org/10.1182/blood-2011-06-356261 [ Links ]
21. Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015;125(19):2908-2914. https://doi.org/10.1182/blood-2015-01-551622 [ Links ]
22. Janka G, Imashuku S, Elinder G, et al. Infection- and malignancy-associated hemophagocytic syndromes: Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12(2):435-444. https://doi.org/10.1016/S0889-8588(05)70521-9 [ Links ]
23. Kasahara Y, Yachie A. Cell type specific infection of Epstein-Barr virus (EBV) in ABV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Blood 2002;44(3);283-294. https://doi.org/10.1016/S1040-8428(02)00119-1 [ Links ]
24. National Institute of Communicable Diseases (NICD). Results of the Fifth South African National HIV Prevalence, Incidence, Behaviour and Communication Survey (SABSSM V) 2017. Communicable Diseases Communiqué 2018;17(8):11. http://www.nicd.ac.za/wp-content/uploads/2018/08/Results-of-the-Fifth-South-African-national-HIV-Prevalence-Incidence-Behaviour-and-Communication-Survey-SABSSM-V-2017.pdf (accessed 29 July 2019). [ Links ]
25. Manji F, Wilson E, Mahe E, Gill J, Conly J. Acute HIV infection presenting as hemophagocytic lymphohistiocytosis: Case report and review of the literature. BMC Infect Dis 2017;17(1):633. https://doi.org/10.1186/s12879-017-2732-y [ Links ]
26. Loganantharaj N, Oliver B, Smith T, Jetly A, Engel L, Sanne S. Hemophagocytic lymphohistiocytosis in an HIV-positive patient with concomitant disseminated histoplasmosis. Int J STD AIDS 2018;29(9):925-926. https://doi.org/10.1177/0956462417753008 [ Links ]
27. Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol 2013;161(5):609-622. https://doi.org/10.1111/bjh.12293 [ Links ]
28. Hejblum G, Lambotte O, Galicier L, et al. A web-based Delphi study for eliciting helpful criteria in the positive diagnosis of hemophagocytic syndrome in adult patients. PLoS ONE 2014;9(4):e94024. https://doi.org/10.1371/journal.pone.0094024 [ Links ]
29. Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program 2009;1:127-131. https://doi.org/10.1182/asheducation-2009.1.127 [ Links ]
30. Akenroye AT, Madan N, Mohammadi F, Leider J. Hemophagocytic lymphohistiocytosis mimics many common conditions: Case series and review of literature. Eur Ann Allergy Clin Immunol 2017;49(1):31-41. https://pdfs.semanticscholar.org/cf22/494a2dad445e22bfc6460face88951dc1e53.pdf (accessed 29 August 2019). [ Links ]
 Correspondence:
Correspondence:
M Nienkemper
mnienk@gmail.com
Accepted 24 February 2020
Contribution of the study. HLH is a rare, life-threatening condition that may be missed in the intensive care setting. This report emphasises the importance of clinical suspicion, early diagnosis and appropriate intervention.


{kind=link}