










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkJournal of the South African Veterinary Association
versão On-line ISSN 2224-9435
versão impressa ISSN 1019-9128
J. S. Afr. Vet. Assoc. vol.93 no.1 Pretoria 2022
http://dx.doi.org/10.36303/jsava.2022.93.1.498
ORIGINAL RESEARCH
https://doi.org/10.36303/jsava.2022.93.1.498
Nemaline myopathy in a six-month-old Pomeranian dog
EG BesterI; AM KitshoffII; WJ BothaIII; E van WilpeIV; L du PlessisV; J WilliamsVI
IDepartment of Companion Animal Clinical Studies, Faculty of Veterinary Science, University of Pretoria, South Africa
IIDepartment of Companion Animal Clinical Studies, Faculty of Veterinary Science, University of Pretoria, South Africa
IIIDepartment of Small Animal Medicine Clinic, Panorama Veterinary Clinic and Specialist Centre, South Africa
IVLaboratory for Microscopy and Microanalysis, Faculty of Natural and Agricultural Sciences, University of Pretoria, South Africa
VElectron Microscope Unit, Department of Anatomy and Physiology, Faculty of Veterinary Science, University of Pretoria, South Africa
VISection of Pathology, Department of Paraclinical Sciences, Faculty of Veterinary Science, University of Pretoria, South Africa
ABSTRACT
Nemaline myopathy - a clinically and genetically complex heterogenous group of disorders - is described uncommonly in humans and rarely in animals, and is characterised by progressive muscle weakness. The diagnosis is confirmed by histological and/or ultrastructural identification of subsarcolemmal, thread-like, rod-shaped structures called nemaline rod bodies within more than 40% of skeletal muscle fibres. These rods contain the Z-line protein, α-actinin, that can be effectively stained in skeletal muscles using Gomori or Masson trichrome and negatively stained with periodic acid-Schiff. Similar rod-like bodies have been found in smaller numbers in dogs with endocrine disorders and occasionally in other conditions in humans. This report is of a six-month-old Pomeranian dog which had progressive exercise intolerance over a two-month period associated with severe disuse muscle atrophy of the thoracic limbs, as well as gradual pelvic limb weakness and regurgitation of food. Baseline diagnostics ruled out endocrinopathies and after histological and ultrastructural evaluation of thoracic limb muscles and nerve biopsies confirmed nemaline myopathy. The clinical course, diagnostic test results, ultrastructure of skeletal muscle and peripheral nerve, gross necropsy findings and histopathology using various stains are described and illustrated.
Keywords: canine, congenital, Gomori trichrome, histopathology, Masson trichrome, nemaline myopathy, ultrastructure
Introduction
Nemaline myopathy (NM) is one of several histologically distinct complex congenital myopathies reported in humans (North et al. 2014). It is rare in animals but has been described in a family of cats (Cooper et al. 1986) and in dogs as an acquired or congenital disorder (Cardinet 1982; Delauche et al. 1998; Dettwiler et al. 2018; Evans et al. 2016; Huxtable et al. 1994). This myopathy comprises a clinically and genetically heterogenous group of disorders, characterised by muscle weakness and the histological identification of thread-like, rod-shaped structures called nemaline rod bodies, within muscle fibres (Shy et al. 1963; North et al. 1997; North et al. 2014). In humans, these rods have been shown in some cases to contain the Z-line protein α-skeletal actinin (Nakamura et al. 2012; Vandebrouck et al. 2010).
Nemaline myopathy is broadly classified in humans as congenital, childhood-onset and adult-onset or late-onset (North & Ryan 1993). Congenital NM is further divided into severe, intermediate and typical, with considerable overlap amongst the various forms (North & Ryan 1993).
The congenital form of NM in humans is characterised by muscle weakness, hypotonia, dysphagia and respiratory difficulty, which, along with recurrent aspiration pneumonia, may be fatal (North et al. 2014). The childhood-onset form of NM occurs in the late first or second decades of life and is slowly progressive. Cardiac muscle involvement with this form of the disorder is rare (North et al. 1997; Ryan et al. 2001). The adult-onset presentation often affects the pelvic limbs, resulting in weakness and protracted clinical symptoms related to an underlying cardiomyopathy (Meier et al. 1984; Stoessl et al. 1985). Other disorders which may
demonstrate a small number of rod bodies in myofibres include human immunodeficiency virus infection (Simpson & Bender 1988; Madonia et al. 2012) polymyositis (Cape et al. 1970) and hypothyroidism (Reyes et al. 1986).
Few cases of NM have been reported in dogs. Of four dogs reported in one study, two had disease unassociated with endocrinopathies and two were associated with concurrent hypothyroidism and hyperadrenocorticism (Delauche et al. 1998). Of the non-endocrine-associated cases, one was a congenital case in a 10-month-old Border Collie and the other an adult-onset case in an 11-year-old Schipperke (Delauche et al. 1998). Congenital NM has also been reported in two 10-week-old sibling female Border Collies, showing progressive gait dysfunction, muscle stiffness and exercise intolerance (Dettwiler et al. 2018), and in a family of cats with onset of clinical signs between six months and 18 months of age (Cooper et al. 1986). Nakamura et al. reported one additional case of adult-onset endocrine-associated NM in a nine-year-old mixed breed dog that presented with generalised skeletal muscle atrophy, atrial standstill and dilation of all cardiac chambers, bacterial cystitis and concurrent primary hypothyroidism (Nakamura et al. 2012). Nemaline rods were found in more than 50% of skeletal muscle fibres diagnosed by modified Gomori trichrome (mGT) stained cryosections, but no rods were found in cardiac muscle biopsies, although specific staining was not done (Nakamura et al. 2012).
In vitro, nemaline rods have been found to originate either due to genetic defects of muscle thin filament such as actin, or due to depletion of adenosine triphosphate in mitochondrial myopathies (Vandebrouck et al. 2010).
In humans, ten genes have been specifically associated with NM: α-skeletal actin (ACTA1); muscle-specific cofilin (CFL2); nebulin (NEB); slow troponin T (TNNT1); fast troponin I (TNNI2); β-tropomyosin (TPM2); slow α-tropomyosin (TPM3); kelch-like family member 40 (KLHL40); kelch-like family member 41 (KLHL41) and leiomodin-3 (LMOD-3) (North et al. 2014; De Winter & Ottenheijm 2017). Muscle-specific ubiquitin ligase (KBTBDI3) can cause both core-rod myopathy and NM with about half of the latter being proposed to be as a result of nebulin mutations due to autosomal recessive disease (North et al. 2014). The condition is genetically complex and may arise from mutations in more than one gene; mutations in one gene leading to different muscle pathologies which include NM; different pathological features in members of a single family or even in one individual at different life stages (North et al. 2014). The mutations of the various genes mentioned previously are present in varying incidence in human NM patients. Some mutations may result in autosomal dominant, sporadic (new dominant), or, in others, autosomal recessive disease, with variable degrees of severity of NM (North et al. 2014; Sandaradura & North 2015).
In a recent study by Evans et al. (2016), a nonsense mutation in NEB (g.52734272 C>A, S8042X) was identified with whole exome sequencing of the dam, two affected littermates, and an unaffected littermate in a family of line-bred American bulldogs with confirmed rod bodies in muscle biopsies (Evans et al. 2016). This study presented the first animal model of NM to be characterised by molecular technique. In this report (Evans et al. 2016), the mutation in the clinically unaffected dam and some of the littermates (affected and unaffected by disease), also suggested recent occurrence of the mutation (Evans et al. 2016). The dam and sire were cousins and were likely heterozygous for the NEB gene, with the affected pups being homozygous due to autosomal recessive inheritance. The female and male known affected littermates had presented at age five months with fairly non-progressive generalised muscle weakness and atrophy beginning at two months, but could still walk. Serum creatine kinase was mildly elevated, a mild decrease in latency of the tibial and ulnar nerves was seen, and there were no respiratory signs. In this study done by Evans et al. (2016) spontaneous electrical fibrillation potentials were recorded electromyographically, within the proximal appendicular muscles of the thoracic limbs and the cervical paraspinal musculature (Evans et al. 2016). Similar NEB nonsense mutation was not found in 100 unrelated American bulldogs nor in 120 dogs of 24 other dog breeds (Evans et al. 2016).
Diagnosis of nemaline rod myopathy includes examination of mGT stained muscle cryosections, muscle immunohisto-chemistry for α-actinin, or ultrastructural examination of muscle by electron microscopy (Nakamura et al. 2012). Rod bodies must occur in more than 40% of muscle fibres for a positive diagnosis of NM (Reyes et al. 1986). Staining of fresh frozen skeletal muscle cryosections by the mGT technique classically demonstrates red or purple nemaline rods in the muscle tissues against a blue-green myofibrillar background (Engel & Cunningham 1963), as well as cytoplasmic inclusion bodies. In formalin-fixed paraffin-wax embedded sections, the staining results of mGM are similar to the Masson trichrome method, but with needing less manipulation and with a cleaner background (Garvey et al. 1996). The rods are not visible or are poorly-apparent with hematoxylin and eosin stain, myofibrillar adenosine tri-phosphatase (ATPase), phosphorylase, or oxidative enzyme reactions (Delauche et al. 1998). The rods stain negatively with periodic acid-Schiff (PAS) (Fukuhara et al. 1978). Ultrastructurally, an accumulation of rods is apparent, with localised enlargement and streaming of the Z lines: the rods appear to originate from the Z disks (Gonatas 1966; Gonatas et al. 1966).
Case presentation
A six-month-old intact male Pomeranian was presented with slowly progressive, non-painful, bilateral, thoracic limb lameness with flexion of the elbow joints over a period of two months. Additionally, general weakness, intermittent regurgitation and severe exercise intolerance that had been getting progressively worse over the preceding weeks were noted. The patient had been treated with anti-inflammatory drugs (drugs used and doses not recorded), physiotherapy and splinted bandages to prevent muscle contracture.
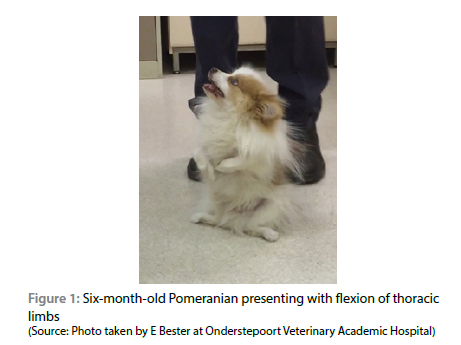
On clinical examination, the dog seemed clinically healthy but was stunted, with flexion of the thoracic limbs, marked thoracic limb muscle atrophy and abducted elbows (begging position) (Figure 1). During manual manipulation, the thoracic limbs could be extended, after which the dog could bear full weight and was able to walk, albeit with a stiff and stilted gait in all four limbs. The patient would easily tire and, after a short period of sternal recumbency, would exhibit recurrent flexion of the elbow joint, with the inability to bear weight on the thoracic limbs. The hocks were in plantigrade stance and adducted when the patient was in a standing position. Orthopaedic examination was all within normal limits. Neurological examination indicated normal cranial nerve reflexes with the exception of the pharyngeal reflex which was abnormal due to the lack of gagging when a finger was introduced down the throat. All the myotactic reflexes were depressed and there was absence of conscious proprioception in the thoracic and pelvic limbs. Decreased withdrawal reflexes were noted in the thoracic limbs. The remainder of the neurological examination was normal. Based on the clinical examination, a neuromuscular disorder was suspected. The breeder of the dog was contacted and indicated that no littermates of this dog had been reported as having similar signs, and neither had any of the breeding stock shown similar clinical signs at any stage up to the time of enquiry.
Complete blood count (ADVIA 2120, Siemens) did not reveal any abnormalities. Serum biochemistry (Cobus Integra 400 Plus, Roche) showed mild elevation of alkaline phosphatase (ALP) (243 U/L; reference interval 20-165 U/L) and alanine aminotransferase (ALT) (84.4 U/L; reference interval 9-73 U/L) activity. The elevated ALP was considered normal in this case due to the young age of the patient. Electrolytes were within their reference ranges. Thoracic radiographs revealed a megaoeosphagus (Figure 2) whereas no orthopaedic abnormalities were detected on thoracic limb radiographs.

The presence of megaoesophagus, with muscle weakness and atrophy, prompted further investigation of muscle, adrenal and thyroid gland functions. Serum creatinine kinase was normal (216 U/L; reference interval 11-254 U/L). A negative neostigmine (Prostigmine®, Canada) response test using 0.02 mg/kg intravenously ruled out myasthenia gravis. Total thyroxine (20.7 nmol/L; reference interval 13-45 nmol/L) and endogenous thyroi-stimulating hormone levels (0.161 ng/mL; reference interval 0.05-0.45 nmol/L) were within reference intervals.
An adrenocorticotrophic hormone (ACTH) stimulation test ruled out muscle atrophy due to hyperadrenocorticism and muscle weakness due to hypoadrenocorticism (basal cortisol 11.2 nmol/L: reference interval 20-200 nmol/L; cortisol one-hour post stimulation 389 nmol/L).
Based on the above-mentioned results, the differential diagnoses included myopathies, neuropathies or neuromyopathies.
Muscle and nerve biopsies were taken from the lateral head of the left m. triceps brachii and n. ulnaris respectively. Samples were preserved in 10% formalin for histopathology and in 2.5% glutaraldehyde for electron microscopy (EM).
Haematoxylin and eosin (H&E) (Bancroft & Gamble2002), periodic acid-Schiff (PAS) (Bancroft & Gamble 2002), Masson trichrome (Bancroft & Gamble 2002; Prophet & Armed Forces Institute of Pathology 1992) and modified Gomori trichrome stains (Ellis, n.d.) were performed on formalin-fixed paraffin wax-embedded biopsied muscle.
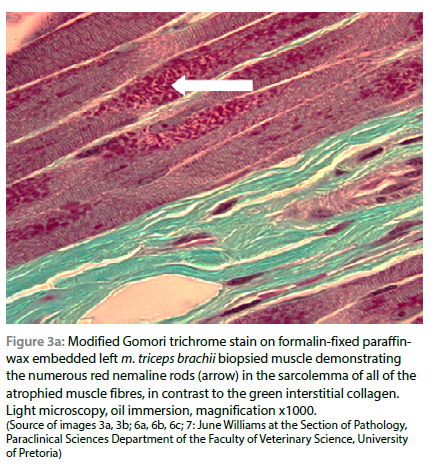
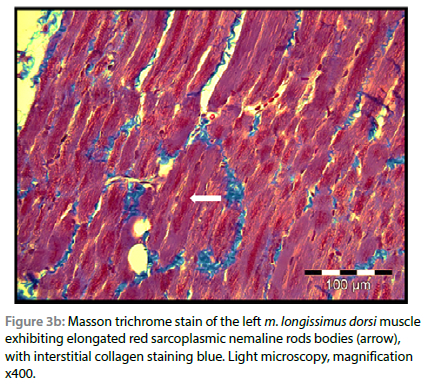
On light microscopic examination of the biopsied m. triceps brachii, using routine H&E staining, all myofibres were atrophied, and numerous clusters of small red-staining rods were seen multifocally in the sarcoplasm of all fibres, especially below the sarcolemma. These rods stained deep red with both trichrome stain variations, but with interstitial collagen fibres staining green with the Gomori method (modified for formalin-fixed tissue) and blue with Masson (Figure 3a and 3b). The rods were visible but less conspicuous using H&E stain. The muscle fibres were uniformly pale-staining with PAS compared with a normal control muscle stained simultaneously, suggesting that they were mostly of type I or glycogen depleted, and the rods stained negatively. There were no visible lesions seen in the ulnar nerve using light microscopy. No histological evidence of Neospora caninum or Toxoplasma gondii infection, nor evidence of inflammation were found. On the basis of the presence of > 40% typical rods in the lateral head of the m. triceps brachii and the clinical findings, a diagnosis of NM was made.
Electron microscopy of the glutaraldehyde-preserved tissues was done using a standard preparation method. Briefly, the tissue was post-fixed in 1% osmium tetroxide, rinsed in Millonig's buffer, dehydrated in a graded series of ethanol, and embedded in a propylene oxide/resin mixture. Ultrathin sections were stained with uranyl acetate and lead citrate prior to examination at 80kV (Graham & Orenstein 2007). The EM (Philips CM10 transmission electron microscope) findings of the biopsied muscle revealed disruption of the Z-lines in the muscle fibers (Figures 4a and 4b). Marked axonal degeneration with dissociation of the myelin sheath surrounding the axons and "blebbing" of myelin into the axonal space of the biopsied n. ulnaris were also seen (Figure 5).


Trial therapy using 2 mg/kg of pyridostigmine (Mestinon, United States) administered orally every eight hours was started, due to anecdotal evidence for the use of cholinesterase inhibitors in the symptomatic management of suspected junc-tionopathies in human cases with congenital myopathies and polyradiculoneuritis (Rodríguez Cruz et al. 2014) and the possibility of false-negative results with the neostigmine response tests. Due to aspiration pneumonia and continued muscle weakness, the owners elected to humanely euthanise the dog one week after initiation of the therapy.
A necropsy revealed marked bilaterally symmetrical muscle atrophy of the thoracic limbs, milder atrophy of other skeletal muscles including the masseter muscles of the head which had a light tan-pink colour and degeneration of the m. longissimus dorsi, and dorsal neck and flank muscles, which had a uniformly pale tan colour. There was a markedly dilated food-filled intrathoracic distal oesophagus (megaoesophagus).
The limbs were detached in toto from the trunk and fixed in a large container of 10% formalin after some longitudinal incisions were made in bulkier muscles to facilitate formalin entry. This was done in order to preserve muscles in situ, prevent contraction as well as to allow future specific muscles to be identified, examined and compared histologically. The head, neck and trunk were similarly preserved as intact as possible. Formalin-fixed tissues of the oesophagus, cardiac muscle, tongue, diaphragm, m. obliquus externus abdominis of the flank, m. longissimus dorsi, dorsal neck region of the m. brachiocephalicus, m. masseters, left and right m. triceps brachii and m. biceps brachii muscles, m. extensor carpi radialis, m. biceps femoris, m. semimembranosus, m. semitendinosus and m. tibialis cranialis were sectioned and stained with H&E and Masson's trichrome stains and evaluated by light microscopy. Some muscle sections and organs were also stained with PAS.
Multifocal clusters of subsarcolemmal nemaline rods were dense and numerous in all fibres of the muscles of the trunk, neck, diaphragm and proximal thoracic limbs when compared with those in pelvic limb muscles and tongue where they were less prevalent. Nemaline rods were scarce in the skeletal muscle of the oesophagus and not seen in the myocardium. Multifocal areas of the m. biceps brachii showed marked muscle atrophy, increase in satellite nuclei, variable fat replacement and occasional destruction and regeneration of fibres (especially a focal mid-lumbar region of m. longissimus dorsi), with little or no inflammatory cell presence.
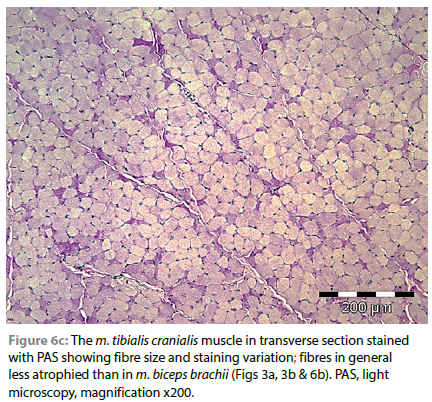
Fibres of the m. tibialis cranialis, which muscles were macro-scopically only mildly atrophic when compared with muscles of the thoracic limbs, with PAS staining, revealed a predominance of pale or intermediate staining fibres, and very few scattered intensely stained fibres (Figure 6a-6c).


Most visceral organs, as well as brain and spinal cord, were also sampled in formalin and examined histologically. The left thyroid gland was macroscopically smaller than the right, both were pale tan, and showed more than 50% empty follicles and variable colloid filling of others. There was very little vacuole presence in cytoplasm of follicular epithelial cells or of colloid at the epithelial interface and no obvious glycogen granules in the follicular apical cytoplasm on PAS staining. Spinal nerves and all brain and spinal cord sections showed no obvious pathology on light microscopy. There was diffuse hydropic vacuolation of hepatocytes in the liver, which stained intensely with PAS, indicating hepatic glycogenosis. The adrenal cortices were neither grossly nor microscopically hypertrophic and cortical cells in the zona fasciculata had vacuolar cytoplasm indicating activity.
Electron microscopical analysis (Jeol JEM2100F Transmission Electron Microscope) was performed on formalinised necropsy samples of the oesophagus, m. biceps brachii, diaphragm, m. longissimus dorsi, m. obliquus externus abdominis, tongue muscles and n. ulnaris (Figures 7a and 7b). The muscles showed typical nemaline rods, consisting of elongated, ovoid and pleomorphic electron-dense deposits replacing the thin filaments in sarcomeres in all the samples submitted, with the oesophageal muscle being the least affected (Figure 8). Areas of relatively normal sarcomeric structure, with normal Z lines, were seen next to disorganised regions containing nemaline rods. Cross striations could be discerned within nemaline rods at high magnification. Lipid droplets were additionally seen in the flank muscle sample. The n. ulnaris sampled during necropsy infrequently demonstrated both unmyelinated and myelinated nerve fibres. The myelin abnormalities noted included the formation of myelin loops and vesicular structures closely associated with the adaxonal surfaces. These changes were similar to those observed in the samples collected antemortally.


Discussion
This report describes a dog with suspected congenital progressive NM and megaoesophagus with no other associated disorders found after extensive testing for various differential diagnoses. Although the dog in this report falls into the congenital form based on the human classification of NM, the progression of clinical signs was rapid in onset, unlike that in humans. Cardiac abnormalities have not been reported in people in this form of NM, and were also absent in this case. In human medicine, as was found in the current dog, the trunk and thoracic limb muscles are most commonly affected. Involvement of the pelvic limb muscles and facial muscles have also been reported, as they were in the current case. In human postmortal studies, extensive diaphragmatic involvement is reported with sparing of cardiac and smooth muscle (Engel et al.1964; Hopkins et al.1966; Kolin 1967; Shafiq et al.1967; Tsujihata et al. 1983), as were similarly found in this dog.
Infants with NM have been described to develop gastro-oesophageal reflux (Berezin et al. 1988). The previously reported family of cats with NM resembled the human adult-onset form as the clinical signs only presented between six months and 1.5 years of age (Cooper et al. 1986). In the case of this report, only a few nemaline rods were found ultrastructurally in the skeletal muscle fibres of the oesophagus. How or whether this contributed to the megaoesophagus is uncertain, but a primary concurrent congenital megaoesophagus is less likely considering the breed of dog.
The relationship between the rod bodies and neuromuscular weakness in NM is unknown because the severity of the weakness does not correlate with the number of rods within the myofibres (Nienhuis et al.1967), although in the current case, it appeared that 100% ofthe fibres of the skeletal muscles examined were affected, each with many clusters of rods and no inflammation, with the least number of rods being observedin the oesophagus and none in the myoca rdium.
In ahdition,this case investigation demonstrated that routinely performed Masson trichrome staining offormalin-fixed muscle is ah accentable anh easier alternative to modified Gomori trichrome staining oS frozen tissues for nemaline rods, hnd as informatiw as mGT staining of Pormalin-fixed tissne,with equivalentimage quality nnd demonstration of the nemaline rods in skeletal muscle.
Neuronal nhanges have been describee in some human cases of NM (Fuku hara et al. 1978) . Recent reports of severa l ty pes of congenitalmyo pnthieswith secondary neuromuscular junction abnormalities suggest that some of these clinical signs seen in human patients may be related to neuromuscularjunction defects (Rodríguez Cruz et al. 2014). This was supported by the positive clinical response in three human patients with centronuclear myopathies treated with pyridostigmine (Rodríguez Cruz et al. 2014). Interpretation of the n. ulnaris findings observed on EM and repeated in the nerves found in the m. triceps brachii muscle of the dog in this case report is uncertain, however the myelin loops suggest either myelin recycling, an early response to axonal atrophy or indicating the presence of redundant myelin (Okasha 2016).
Nerve lesions including narrowed axon diameters and myotendinous junction-like structures as well as fibre splitting were found in three human siblings (Fukuhara et al. 1978). The latter two lesions were often found near each other but could be differentiated from each other because the myotendinous junction-like structures showed sarcolemma thickening not seen in fibre splitting lesions. These authors suggested nemaline rods may result from longitudinal splitting and disruption of fibres subsequent to deficient fibre regeneration due to possible neurotrophic abnormalities (Fukuhara et al. 1978). In the current case, these changes were not visible on light microscopic examination of nerves found amongst biopsy and necropsy muscle sections, and neuromuscular junctions were not found in the EM samples examined. Neuropathy has been recorded in hypothyroid patients (North et al. 1997), however the current dog was not typically hypothyroid on specific endocrine testing but did show histological follicular atrophy and quiescence, which may have been due to endogenous cortisol suppression related to stress.
Rod bodies have been described in other pathological and experimental conditions suggesting that they are not specific only for congenital nemaline myopathy. Rods have also been associated with polymyositis (Cape et al. 1970), spinal muscle atrophy (Konno et al 1987), rhabdomyoma, HIV infections (Delauche et al. 1998; Madonia et al. 2012) and an adult with primary hypothyroidism (Reyes et al. 1986) in human literature. In these instances the rods were too few in number to allow classification as nemaline myopathy, for which more than 40% of muscle fibres should be affected (Reyes et al. 1986). Rods have also been associated in a Labrador Retriever with generalised muscle wasting and an adenocarcinoma of the small intestine (Cardinet & Holliday 1979). In humans, a severe form of NM with shortened lifespan, significant forearm and isolated muscle weakness, decreased mobility, fibre atrophy and an increase in slow fibres is a result of a normal allele of the alpha-skeletal actin gene replaced with a mutated form (H40Y) (Nguyen et al. 2011). The dog of the current report presented similarly, and in the absence of specific genetic testing, it could be speculated that the animal may have had a similar genetic defect. Mice from a mouse model of severe NM generated by replacing a normal allele of the alpha-skeletal actin gene with the mutated H40Y gene, showed alleviation of their mobility defect when supplemented with dietary L-tyrosine. L-tyrosine supplementation also showed an improvement in skeletal muscle pathologies, a reduction in chronic myofibre repair and a reduction in nemaline rods numbers (Nguyen et al. 2011). In future canine cases with similar presentation, this L-tyrosine nutritional supplementation could be attempted as a treatment option.
This NM mouse model (Nguyen et al. 2011) showed muscle lesions including nemaline rods, fibre atrophy and increase in slow fibres, as typically seen in human patients with NM. In the current dog case, affected muscles showed a predominance of fibres with pale PAS staining suggesting low glycogen content and possibly representing a predominance of type 1 or slow fibres, while there was marked glycogenosis in hepatocytes; this latter may, however, have been influenced by stress-related endogenous cortisol.
Although the mechanism of rod formation and the muscle weakness is not fully understood, it is important for clinicians to recognise NM as a differential for muscle weakness and atrophy in dogs, with the aim of diagnosis and further investigation in order to better understand the genetics, pathophysiology and the pathogenesis of this disorder.
Animal ethics for case publication
The current case involves the description of the clinical findings, routine work up and case management of a patient. The University of Pretoria Animal Ethics Committee approved the handling of and writing up of this case in retrospect .
Conflict of interest
The authors declare no conflict of itnerest.
Funding source
None.
Ethical approval
The current case involves the description of the clinical findings, routine work up and case management of a patient. The University of Pretoria Animal Ethics Committee approved the handling of and writing up of this case in retrospect (Ref V019-18).
ORCID
EG Bester https://orcid.org/0000-0003-0471-3326
AM Kitshoff https://orcid.org/0000-0001-9657-740X
WJ Botha https://orcid.org/0000-0002-5774-0320
Ε van Wilpe https://orcid.org/0000-0001-9410-1221
L du Plessis https://orcid.org/0000-0002-1330-1334
References
Bancroft, J.D., Gamble, M., 2002, Theory and practice of histological techniques, Churchill Livingstone, Elsevier Science Limited. [ Links ]
Berezin, S., Newman, L.J., Schwarz, S.M., Spiro, A.J., 1988, Gastroesophageal reflux associated with nemaline myopathy of infancy, Pediatrics 81(1), 111-115. https://doi.org/10.1542/peds.81.1.111. [ Links ]
Cape, C.A., Johnson, W.W., Pitner, S.E., 1970, Nemaline structures in polymyositis. A nonspecific pathological reaction of skeletal muscles, Neurology 20(5), 494-502. https://doi.org/10.1212/WNL.20.5.494. [ Links ]
Cardinet, G.H., 1982, Nemaline rods in neuromuscular disorders of the dog, 119th Annual Meeting, American Association of Veterinary Anatomists. [ Links ]
Cardinet, G.H., Holliday, T.A., 1979, Neuromuscular diseases of domestic animals: A summary of muscle biopsies from 159 cases, Ann N Y Acad Sci 317, 290-313. https://doi.org/10.1111/j.1749-6632.1979.tb56538.x. [ Links ]
Cooper, B.J., De Lahunta, A., Gallagher, E.A.,Valentine, B.A., 1986, Nemaline myopathy of cats, Muscle Nerve 9(7), 618-625. https://doi.org/10.1002/mus.880090707. [ Links ]
Dettwiler, M., Sydler, T., Klausmann, S., et al., 2018, Nemaline myopathy diagnosed in two young Border collies in formalin-fixed paraffin-embedded muscle samples using conventional stains, Vet Rec 6, e00711. https://doi.org/10.1136/vetreccr-2018-000711. [ Links ]
De Winter, J.M., Ottenheijm, C.A.C., 2017, Sarcomere dysfunction in nemaline myopathy, J Neuromuscul Dis 4(2), 99-113. https://doi.org/10.3233/JND-160200. [ Links ]
Delauche, A.J., Cuddon, P.A., Podell, M., et al., 1998, Nemaline rods in canine myopathies: 4 case reports and literature review, J Vet Intern Med 12(6), 424-430. https://doi.org/10.1111/j.1939-1676.1998.tb02145.x. [ Links ]
Ellis, R., Gomori's trichrome staining protocol for connective tissues. IHCWORLD, Life Science Products & Services. Available from: http://www.ihcworld.com/_protocols/special_stains/gomori's_trichrome_ellis.htm. [ Links ]
Engel, W.K., Cunningham, G.G., 1963, Rapid examination of muscle tissue. An improved trichrome method for fresh-frozen biopsy sections, Neurology 13,919-923. https://doi.org/10.1212/WNL.13.11.919. [ Links ]
Engel, W.K., Wanko, T., Fenichel, G.M., 1964, Nemaline myopathy; a second case, Arch Neurol 11(1), 22-39. https://doi.org/10.1001/archneur.1964.00460190026003. [ Links ]
Evans, J.M., Cox, M.L., Huska, J., et al., 2016, Exome sequencing reveals a nebulin nonsense mutation in a dog model of nemaline myopathy, Mamm Genome, 27(9-10), 495-502. https://doi.org/10.1007/s00335-016-9644-9. [ Links ]
Fukuhara, N., Yuasa, T., Tsubaki, T., et al., 1978, Nemaline myopathy: Histological, histochemical and ultrastructural studies, Acta Neuropathol 42(1), 33-41. https://doi.org/10.1007/BF01273264. [ Links ]
Garvey, W., Bigelow, F., Fathi, A., et al., 1996, Modified gomori trichrome stain for frozen skeletal muscle and paraffin embedded sections, Journal of Histotechnology 19(4), 329-333. https://doi.org/10.1179/his.1996.19.4.329. [ Links ]
Gonatas, N.K., 1966, The fine structure of the rod-like bodies in nemaline myopathy and their relation to the z-discs, J Neuropathol Exp Neurol 25(3), 409-421. https://doi.org/10.1097/00005072-196607000-00005. [ Links ]
Gonatas, N.K., Shy, G.M., Godfrey, E.H., 1966, Nemaline myopathy. The origin of nemaline structures N Engl J Med 274(10), 535-539. https://doi.org/10.1056/NEJM196603102741002. [ Links ]
Graham, L., Orenstein, J.M., 2007, Processing tissue and cells for transmission electron microscopy in diagnostic pathology and research, Nature Protocols 2(10), 2439-2450. https://doi.org/10.1038/nprot.2007.304. [ Links ]
Hopkins, I.J., Lindsey, J.R., Ford, F.R., 1966, Nemaline myopathy. A long-term clinicopathologic study of affected mother and daughter, Brain 89(2), 299-310. https://doi.org/10.1093/brain/89.2.299. [ Links ]
Huxtable, C.R., Chadwick, B., Eger, C., Shaw, S. 1994. Severe subacute progressive myopathy in a young silky terrier, Progress in Veterinary Neurology 5(1),21-27. [ Links ]
Kolin, I.S., 1967, Nemaline myopathy. A fatal case, Am J Dis Child 114(1), 95-100. https://doi.org/10.1001/archpedi.1967.02090220101019. [ Links ]
Konno, H., Iwasaki, Y., Yamamoto, T., Inosaka, T., 1987, Nemaline bodies in spinal progressive muscle atrophy: An autopsy case, Acta Neuropathol 74, 84-88. https://doi.org/10.1007/BF00688343. [ Links ]
Madonia, P., Wilson, J., Bican, O., et al., 2012, HIV, rods, and the muscles--a discussion about HIV-associated nemaline rod myopathy, J La State Med Soc 164(6), 320-323. [ Links ]
Meier, C., Voellmy, W., Gertsch, M., et al., 1984, Nemaline myopathy appearing in adults as cardiomyopathy. A clinicopathologic study, Arch Neurol 41(4), 443-445. https://doi.org/10.1001/archneur.1984.04050160109025. [ Links ]
Nakamura, R.K., Russell, N.J., Shelton, G.D., 2012, Adult-onset nemaline myopathy in a dog presenting with persistent atrial standstill and primary hypothyroidism, J Small Anim Pract 53(6), 357-360. https://doi.org/10.1111/j.1748-5827.2012.01221.x. [ Links ]
Nguyen, M.A., Joya, J.E., Kee, A.J., et al., 2011, Hypertrophy and dietary tyrosine ameliorate the phenotypes of a mouse model of severe nemaline myopathy, Brain 134(12), 3516-3529. https://doi.org/10.1093/brain/awr274. [ Links ]
Nienhuis, A.W., Coleman, R.F., Brown, W.J., et al.,1967, Nemaline myopathy. A histopathologic and histochemical study, Am J Clin Pathol 48(1), 1-13. https://doi.org/10.1093/ajcp/48.1.1. [ Links ]
North, K.N., Ryan, M.M., 1993, Nemaline myopathy. In: Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., et al. (eds.) Genereviews®. Seattle (WA): University of Washington, Seattle. [ Links ]
North, K.N., Laing, N.G., Wallgren-Pettersson, C., 1997, Nemaline myopathy: Current concepts. The ENMC International Consortium and nemaline myopathy, J Med Genet 34(9), 705-713. https://doi.org/10.1136/jmg.34.9.705. [ Links ]
North, K.N., Wang, C.H., Clarke, N., et al., 2014, Approach to the diagnosis of congenital myopathies, Neuromuscul Disord 24(2), 97-1 16. https://doi.org/10.1016/j.nmd.2013.11.003. [ Links ]
Okasha, E.F., 2016, Effect of long term-administration of aspartame on the ultrastructure of sciatic nerve, J Microsc Ultrastruct 4, 175-183. https://doi.org/10.1016/j.jmau.2016.02.001. [ Links ]
Prophet, E.B., Armed Forces Institute of Pathology, 1992, Laboratory methods in histotechnology, Washington, D.C., American Registry of Pathology. [ Links ]
Reyes, M.G., Tal, A., Abrahamson, D., Schwartz, M., 1986, Nemaline myopathy in an adult with primary hypothyroidism, Can J Neurol Sci 13(2), 117-119. https://doi.org/10.1017/S0317167100036039. [ Links ]
Rodríguez Cruz, P.M., Sewry, C., Beeson, D., et al., 2014, Congenital myopathies with secondary neuromuscular transmission defects; a case report and review of the literature, Neuromuscul Disord 24(12), 1103-1110. https://doi.org/10.1016/j.nmd.2014.07.005. [ Links ]
Ryan, M.M., Schnell, C., Strickland, C.D., et al., 2001, Nemaline myopathy: A clinical study of 143 cases, Ann Neurol 50(3), 312-320. https://doi.org/10.1002/ana.1080. [ Links ]
Sandaradura, S., North, K.N., 2015, Lmod3: The "missing link" in nemaline myopathy?, Oncotarget 6(29), 26548-26549. https://doi.org/10.18632/oncotarget.5267. [ Links ]
Shafiq, S.A., Dubowitz, V., Peterson, H.E.C., Milhorat, A.T., 1967, Nemaline myopathy: Report of a fatal case, with histochemical and electron microscopic studies, Brain 90(4), 817-828. https://doi.org/10.1093/brain/90.4.817. [ Links ]
Shy, G.M., Engel, W.K., Somers, J.E., Wanko, T., 1963, Nemaline myopathy. A new congenital myopathy, Brain 86(4), 793-810. https://doi.org/10.1093/brain/86.4.793. [ Links ]
Simpson, D.M., Bender, A.N., 1988, Human immunodeficiency virus-associated myopathy: Analysis of 11 patients, Ann Neurol 24(1), 79-84. https://doi.org/10.1002/ana.410240114. [ Links ]
Stoessl, A.J., Hahn, A.F., Malott, D., et al., 1985, Nemaline myopathy with associated cardiomyopathy. Report of clinical and detailed autopsy findings, Arch Neurol 42(11), 1084-1086. https://doi.org/10.1001/archneur.1985.04060100070025. [ Links ]
Tsujihata, M., Shimomura, C., Yoshimura, T., et al., 1983, Fatal neonatal nemaline myopathy: A case report, J Neurol Neurosurg Psychiatry 46(9), 856-859. https://doi.org/10.1136/jnnp.46.9.856. [ Links ]
Vandebrouck, A., Domazetovska, A., Mokbel, N., et al., 2010, In vitro analysis of rod composition and actin dynamics in inherited myopathies, J Neuropathol Exp Neurol 69(5), 429-441. https://doi.org/10.1097/NEN.0b013e3181d892c6. [ Links ]
 Correspondence:
Correspondence:
email: elgebester@gmail.com

