










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkJournal of the South African Veterinary Association
versión On-line ISSN 2224-9435
versión impresa ISSN 1019-9128
J. S. Afr. Vet. Assoc. vol.85 no.1 Pretoria ene. 2014
CASE REPORT
L-2 hydroxyglutaric aciduria in a South African Staffordshire Bull Terrier
Marlies BóhmI; Howard HendersonII; Henriette van der ZwanIII; Sandra BassonIV
IKing Edward Veterinary Referral Hospital, Port Elizabeth, South Africa
IIUCT/NHLS Department of Chemical Pathology, South Africa
IIIInqaba biotechnical industries (Pty) Ltd., South Africa
IVDrs Visser, Erasmus, Vawda & Partners, Port Elizabeth, South Africa
ABSTRACT
L-2 hydroxyglutaric aciduria is an autosomal recessive error of metabolism that manifests as an encephalopathy. The most common presenting signs are seizures, tremors, ataxia and/ or dementia. Some affected dogs show only subtle behavioural changes. Amongst canines, the condition has been best described in Staffordshire Bull Terriers. Although this is the first reported case in South Africa, at least three other affected dogs have been indentified by polmerase chain reaction (PCR) in this country. Affected dogs have normal haematology, serum biochemistry and routine urine analysis. This report discusses the advantages and limitations of the three main diagnostic modalities, namely: magnetic resonance imaging, urine gas chromatography-mass spectrometry and genetic testing. The aim of this report is to increase awareness of the condition, assist diagnosis in encephalopathic dogs and improve detection of carriers amongst breeding stock.
Introduction
L-2 hydroxyglutaric aciduria (L2-HGAU) was first reported in humans in 1980 (Duran et al. 1980). To date, case reports on well over 100 patients have been published (Kranendijk et al. 2012; Steenweg et al. 2010). In 2003, Abramson and others described six-affected Staffordshire Bull Terriers (SBT) in the United Kingdom (Abramson et al. 2003). Since then, L2-HGAU has also been documented in three Yorkshire Terriers (YRT) (Farias et al. 2012; Sanchez-Masian et al. 2012) and one West Highland White Terrier (Garosi et al. 2005). The causative mutations in YRT differ from those in SBT (Farias et al. 2012; Penderis et al. 2007; Sanchez-Masian et al. 2012). Whilst all tested SBT have had the same mutation in the L2 hydroxyglutarate dehydrogenase gene (L2-HGDH) (Penderis et al. 2007), over 80 different mutations of L2-HGDH have been demonstrated in affected humans (Kranendijk et al. 2012; Steenweg et al. 2010).
Case history
A 5-year-old, 18.3 kg, female, neutered SBT was referred to the first author for further investigation of an episode of bizarre behaviour or dementia.
In retrospect, her owners had observed mild ataxia for 11 months prior to presentation; she appeared somewhat unsteady for a few seconds when she woke up, occasionally banged into walls on cornering and at times had difficulty climbing steps. Signs would wax and wane and were too subtle to prompt consultation with a veterinarian.
The first episode of bizarre behaviour occurred nine months prior to presentation, lasted for a day and resolved without treatment. The dog appeared disorientated and aggressive, was pacing and restless, panting and hyperactive. She did not respond to commands where she was normally biddable.
The episode that prompted the investigation lasted for a week. Initially, she became unusually aggressive toward the neighbour's dog and attacked the garbage bins. Signs gradually worsened and two days later she had stopped responding to commands, spent all night pacing and appeared disorientated with impaired spatial perception and balance. She urinated indoors and showed no awareness of the fact that this had happened. On the third symptomatic day she was presented to her usual veterinarian. She was treated with alprazolam (1.75 mg bid) (Adco-Alzem, Adcock Ingram Pharmaceuticals, Bryanston, South Africa), which caused transient sedation, after which the above behaviour resumed. The next day procaine benzylpenicillin with dihydrostreptomycin (dose not recorded) (Depomycin, Intervet-Schering Plough, Isando, South Africa), alprazolam, dexamethazone (dose not recorded) (Dexa 0.2 Phenix, Virbac, Halfway House, South Africa) and phenobarbitone (30 mg bid) (Lethyl, Aspen Pharmacare, Gallo Manor, South Africa) were administered. Her owners reported that she had appeared calmer and had slept for the first time in two days following these treatments. She was referred to a specialist the following day, which was Day 5 of this episode.
On clinical examination the dog appeared disorientated. She was staring into space, becoming fixated on and staring at objects (e.g. a lead), pacing, circling, and walking into a wall. A complete clinical and neurological examination revealed no further abnormalities.
Results of haematology, serum biochemistry, electrolytes, random cortisol, basal ammonia and a bile acid stimulation test were all within normal reference ranges. Urine analysis revealed only hyposthenuria (SG 1.005).
Anaesthesia was induced with intravenous propofol at 3.33 mg/kg (Propofol 1.0% Fresenius, Fresenius Kabi, Halfway House, South Africa) and was maintained with a constant rate infusion of propofol 0.20 mg/kg/min in saline (5 mL/kg/min) (Sabax Sodium Chloride 0.9%, Adcock Ingram, Johannesburg, South Africa). The following MRI sequences were acquired using an eight-channel head array coil in a GE 1.5T Sigma Excite HD MRI scanner: transverse T2, transverse T2 FLAIR, sagittal T1, sagittal T2 and coronal T2 FLAIR and axial T1. Further sagittal and transverse T1 sequences were acquired after intravenous administration of 1.8 mmol gadolinium (Dotarem, Gd-DOTA 27.93% m/v, 0.5 mmol/mL, Guerbet, Villepinte, France). The MRI was interpreted by one of the co-authors (SB). A subtle bilateral symmetrical T2 hyperintensity was evident, involving the regions of the basal ganglia, central and posterior tectum. No contrast enhancement was evident and no other abnormalities were reported. MRI changes were consistent with a metabolic or toxic encephalopathy.
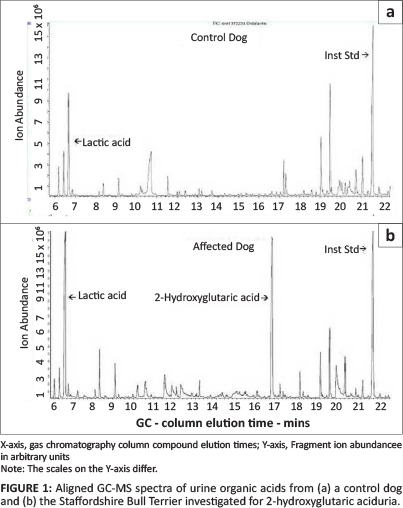
A single free flow urine sample was collected from the patient and submitted to the UCT/NHLS laboratory (Red Cross Children's Hospital, Cape Town) for organic acid analysis by gas chromatography-mass spectrometry (GC-MS) as previously described (Van der Watt et al. 2010). Urine from a clinically normal five-year-old female entire Doberman bitch was used as a control. The organic acid spectrum from the patient revealed significant lactic aciduria and a large 2-hydroxyglutaric acid component (quantified at 360 mmol/mol creatinine). 2-Hydroxyglutaric acid was not detected in the control canine urine (Figure 1). The patient's urine organic acid profile was highly suggestive of L2-HGAU, as previously reported in SBTs (Abramson et al. 2003).
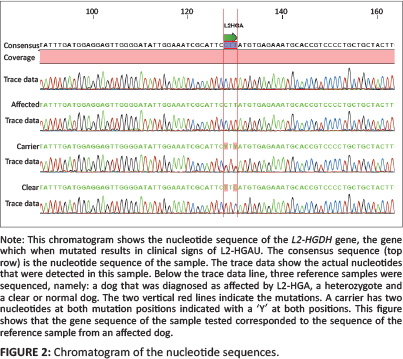
EDTA blood was submitted to Inqaba Biotec (Hatfield, Pretoria, South Africa) to determine whether the causative mutation was present. DNA was extracted and followed with a polymerase chain reaction (PCR) to amplify the gene of interest. The causative mutation is known to be in exon 10 of chromosome 8 in SBT and consists of two single-nucleotide substitutions separated by a single T nucleotide (c[1297TRC; 1299cRt]). This results in the substitution of two adjacent amino acids: proline replaces leucine at position 433 and tyrosine replaces histidine at position 434 (Penderis et al. 2007). An ABI PRISM™ 3130 Genetic Analyzer was used to determine the nucleotide sequences in the amplicons. The dog was shown to be homozygous for the dicodon mutation in L2-HGDH (Figure 2).
The patient was maintained on low doses of phenobarbitone (30 mg in the morning and 45 mg in the evening; serum phenobarbitone 49.8 µmol/L - therapeutic range 43 µmol/L -172 µmol/L) until she was relocated with her owners. On the three occasions when the first author observed the patient in the three months after her initial diagnosis she still appeared mildly dazed. The dose was reduced to 15 mg in the morning and 30 mg in the evening at some stage, at the recommendation of another veterinarian. Supplementation with 100 mg riboflavin daily started immediately after diagnosis. Her owners considered her back to normal and no further severe episodes were observed for the remainder of her life. She was treated with both medications until she ruptured bilateral cruciate ligaments, prompting euthanasia 16 months after diagnosis.
Discussion
This is the first report of L2-HGAU in SBT in South Africa. Samples from 200 South African SBT were submitted to Inqaba Biotec, a commercial laboratory, for determination of the dogs' L2-HGDH status between 2009 and 2012. The laboratory detected 46 heterozygotes (23%) and four (2%) affected dogs (H. van der Zwan [Inqaba Biotec] pers. comm., 04 Dec. 2012). This confirms that the mutation is present in the South African SBT population and dogs showing clinical signs of L2-HGAU are likely to be presented periodically to veterinarians.
A study of 130 normal UK SBT and 131 normal Finnish SBT selected from two DNA archives detected L2-HGDH heterozygotes in 11% of both populations (Short et al. 2010). The same authors searched for the SBT L2-HGDH mutations in residual blood samples submitted for routine phenobarbitone monitoring from over 1000 epileptic dogs of other breeds and found no carrier dogs (Short et al. 2010). Thus, the mutations described by Penderis et al. (2007) are either specific to the SBT breed or occur at a very low frequency in epileptic dogs of other breeds. The higher proportion of carriers and affected dogs recorded by Inqaba Biotec is expected because their data merely reflect the incidence of positive samples submitted to a commercial laboratory and do not represent a random sampling of the South African SBT population. Staffordshire Bull Terrier breeders who had become aware of L2-HGAU and were screening their breeding stock submitted most samples. It is not known what proportion of the sampled SBTs was symptomatic or had known, affected relatives. Carrier and affected dogs identified by the laboratory originated from different breeding lines and lived in kennels dispersed throughout South Africa.
L2-HGAU has been called a deficiency of 'metabolic repair'. L2-HGA is produced because the mitochondrial enzyme L-malate dehydrogenase is not specific for its primary substrate, oxaloacetate, but is also able to reduce a-ketoglutarate to L2-hydroxyglutaric acid (L2-HGA). L2-HGA has no known metabolic function. It is converted back to α-ketoglutarate by L2-hydroxyglutaric acid dehydrogenase (L2-HGDH). Thus, L2-HGDH ensures that the functionless L2-HGA does not accumulate in the cells of normal animals, in other words, it ensures metabolic repair (Rzem et al. 2004) (Figure 3).

When a mutated L2-HGDH results in a non-functional enzyme, L2-HGA accumulates. Markedly increased L2-HGA levels can be measured in CSF, plasma and urine in affected individuals (Rzem et al. 2004; Steenweg et al. 2010) but the authors are not aware of any publication proving accumulation of this substance in astrocytes or any other neurons. L2-HGDH is most active in the brain (Sanchez-Masian et al. 2012); this may explain why neurological signs predominate in affected animals. L2-HGA has been shown to cause oxidative stress - it inhibits mitochondrial creatinine k ase in the cerebellum and may also inhibit glutamate-dependent pathways, as it is a structural analogue of this common neurotransmitter substance (Rzem et al. 2004). It is this oxidative stress that was thought to be responsible for the astrocyte vacuolation observed on histopathological sections of an affected dog's brain (Scurrell et al. 2008).
Affected humans usually show clinical signs before they are seven years old (Kranendijk et al. 2012) but, as signs may be subtle and/or non-specific, diagnosis may be delayed as in this case (Abramson et al. 2003; Patay et al. 2012; Weimar et al. 2012). Clinical signs shown by both species include ataxia, dementia and seizures. In dogs, signs of dementia may include head pressing, getting trapped in corners, hyperactivity, aggression, lethargy or attention seeking (Farias et al. 2012). Dogs show behavioural changes and/or loss of learnt behaviour, whilst behavioural problems are reported in 32% and psychomotor delay in 93% of humans. Disorientation, hypermetria and tremors are reported in dogs, whilst 82% of humans specifically show cerebellar ataxia with intention tremors (Steenweg et al. 2010). Thirty-eight percent of people also show extrapyramidal signs, such as involuntary movements, inability to initiate movement, tremor and muscle spasm. Owners have observed muscle stiffness and fatigue with exercise in affected dogs, whilst 40% of human patients show hypotonia progressing to spasticity (Abramson et al. 2003). Some dogs show periodic exacerbation of clinical signs that may last for minutes to days. In contrast, clinical signs are slowly progressive in humans and periodic exacerbations are not commonly reported. Many dogs will show only one or two of the above signs and some may be so subtle that the owner considers the dog to be normal (Abramson et al. 2003). In both species the extent of the neurological compromise does not appear to be related directly to the severity of the aciduria (Abramson et al. 2003). In humans, it has not been possible to associate different mutations with different clinical manifestations (Steenweg et al. 2010).
On MRI, both species show T2 hyperintensity in peripheral subcortical white matter as well as in grey matter of the thalamus, basal ganglia, globus pallidus, caudate and dentate nuclei (Penderis et al. 2007; Steenweg et al. 2010). Diffuse oedema, manifesting as a subtle T1 hypointensity without contrast enhancement, is sometimes reported (Abramson et al. 2003). In addition, humans may show cerebellar atrophy, macrocephaly and may also have an increased risk of brain tumours (Aghili, Zahedi & Rafiee 2009; Kranendijk et al. 2012). To the authors' knowledge, none of these have been reported in dogs. Because the extensive grey matter changes in dogs are usually symmetrical, inexperienced viewers may overlook them. In addition, other breed-associated polioencephalopathies may have similar MRI lesions (Abramson et al. 2003). Referral to a veterinary diagnostic imaging specialist is thus advised.
A full post mortem study of an affected SBT found no macroscopic changes. Histopathological changes were restricted to the brain. There were marked spongiform changes in the astrocytes of the cerebral cortex, thalamus, cerebellum and brainstem. Grey matter was most severely affected (Scurrell et al. 2008). In humans, spongiform changes are most severe in the subcortical white matter (Penderis et al. 2007). Thus, histopathological changes correspond to those found on MRI. It is not clear why the distribution of the most severe lesions differs between humans and dogs (Abramson et al. 2003).
Routine haematology, serum biochemistry and in-house urine analysis are typically normal (Scurrell et al. 2008) and the hyposthenuria in this patient was probably a side effect of phenobarbitone treatment.
Routine GC-MS detects the presence of 2-hydroxyglutaric acid in the urine, but does not determine chirality. L2-HGAU must be differentiated from D-2 hydroxyglutaric aciduria (D2-HGAU), isolated cases of which have been reported in dogs (Abramson et al. 2003). In humans, two types of congenital D2-HGAU, as well as D,L-2 hydroxyglutaric aciduria, have been described (Kranendijk et al. 2012). To complicate matters, in humans, D2-HGAU has been associated with other errors of metabolism, for example, a skeletal dysplasia resulting in metaphyseal chondromatosis and multiple Acyl-CoA dehydrogenase deficiency. Lastly, it has been observed as an acquired mutation in patients with glioma and acute myeloid leukaemia (Kranendijk et al. 2012). There is no reason why these errors of metabolism would not occur in dogs, thus all of these should be considered as a differential diagnosis for a dog with high 2-hydroxyglutaric acid levels in the urine. In humans, clinical signs and MRI changes may often distinguish these conditions, but confirmation of chirality and genetic testing are necessary for a complete work-up (Kranendijk et al. 2012). Urine GC-MS would be an appropriate screening tool for L2-HGAU in encephalopathic dogs of breeds other than SBT. Dogs with a 2-hydroxyglutaric acid spike in their urine may not have L2-HGAU, but the disease can be excluded in dogs without one.
Elevated lysine in the CSF, plasma and urine has been noted in some humans and dogs with L2-HGAU, whilst some had elevated urine cysteine and/or arginine (Abramson et al. 2003). As high urine lysine decreases absorption of cysteine, ornithine and arginine in the proximal convoluted tubules, increased loss of these four amino acids may be interrelated (Abramson et al. 2003). Urine or plasma amino acid levels were not quantified in this case, but are of value in detecting other metabolic encephalopathies, for example, multiple Acyl-CoA dehydrogenase deficiency and maple syrup urine disease (MSUD). Elevated urine lactate has been reported in one person with L2-HGAU (Barth et al. 1998).
More than 80 different mutations of L2-HGDH that can result in a non-functional enzyme and clinical signs of L2-HGAU have been documented in humans (Kranendijk et al. 2012). Such genetic heterogeneity should be anticipated amongst dogs. Indeed, a different L2-HGDH mutation has already been described in YRT (Farias et al. 2012). Genetic screening is by its nature specific to a known mutation and cannot be used to exclude the disease. It is, however, the only way in which carriers can be detected. Genetic screening and GC-MS thus have complimentary roles in the diagnosis of L2-HGAU.
L2-HGAU is incompletely understood and there is no specific treatment for dogs or humans at present (Penderis et al. 2007). Humans with better-characterised organic acidurias are treated with dietary protein restriction and appropriate amino acid, mineral and vitamin supplementation (Kolker et al. 2012; Penderis et al. 2007; Van der Watt et al. 2010). The efficacy of such supplementation of precursors is likely to vary depending on the nature of the underlying mutation as well as whether any normal enzyme is present in the particular patient. Supplementation with 100 mg riboflavin once daily improved neurological function and markedly decreased L2-HGA secretion in the urine of two of three children with L2-HGAU in whom its use is reported (Kranendijk et al. 2012; Yilmaz 2009). Riboflavin (Vitamin B2) is bound to an ADP molecule to form flavin adenine dinucleotide (FAD). Flavin adenine dinucleotide is a co-factor for L2-HGDH (Yilmaz 2009). A diet supplemented with FAD and levocarnitine chloride improved clinical signs in another human (Samuraki et al. 2008). A lysine-restricted diet was offered to one dog with concurrently elevated plasma lysine levels, but the patient refused to eat it (Farias et al. 2012). Such a diet was not tested in this SBT, as her lysine status was not known. In dogs with seizures, phenobarbitone at 3 mg/kg appears effective at controlling clinical signs (Abramson et al. 2003; Sanchez-Masian et al. 2012). In the SBT described here, it is believed that the phenobarbitone acted as a sedative during the period of acute exacerbation that prompted investigation. The dose was reduced to 15 mg in the morning and 30 mg in the evening, but was not discontinued in the hope that it might suppress another period of dementia.
The therapeutic effect of either of the interventions used in this case is difficult to assess in this dog, as her severe manifestations were episodic and uncommon, she only lived for 16 months post diagnosis and she was never off medication again. This case does confirm previous observations that some mildly affected individuals may remain acceptable pets despite the limited scope of current treatment options.
Conclusion
To the authors' knowledge, this is the first report of L2-HGAU in a South African SBT. The aim of this report is to raise awareness of this condition in the SBT breed in South Africa for two reasons. The first is so that colleagues can encourage breeders to screen breeding stock prior to mating. At least 46 carrier dogs have already been identified in South Africa and 11 % of two overseas convenience samples of clinically normal SBT were carriers. Heterozygotes are asymptomatic but unwittingly mating two carriers is likely to result in clinically affected pups. Some affected dogs have clinical signs that are vague and may even be considered a cute quirk or 'just a Staffie thing' by some owners. Nevertheless, even mildly affected individuals should not be bred because all offspring will at least be carriers and subsequent homozygous offspring could be a great deal more compromised. The second reason is so that colleagues consider genetic screening on SBT showing consistent neurological signs, as this potentially avoids having to do more invasive or expensive diagnostic testing. There may be further mutations causing L2-HGAU in dogs, just as there are over 80 described in humans (Kranendijk et al. 2012). For this reason, urine organic acids should be determined in dogs showing consistent clinical signs or MRI changes but whose genetic test suggests they are unaffected.
Acknowledgement
Competing interests
The authors declare that they have no financial or personal relationship(s) which may have inappropriately influenced them in writing this article.
Authors' contributions
M.B. (King Edward Veterinary Referral Hospital) was the primary clinician responsible for looking after the patient, coordinating work-up, writing the main body of the article, performing the literature search and coordinating everyone else. H.H. (UCT/NHLS Department of Chemical Pathology) performed GC-MS on urine, wrote the section on the 2-hydroxyglutaric acid analysis of the urine for the article, performed an extensive review of the article and added further references pertinent to the GC-MS. H.v.d.Z. (Inqaba biotechnical industries [Pty] Ltd.) performed the PCR, supplied data on other Staffordshire Bull Terriers tested, wrote the section on the PCR analysis for the article and reviewed the remainder of the article. S.B. (Drs Visser, Erasmus, Vawda & Partners) reviewed the MRI, checked that the imaging findings were correctly reported in the article and reviewed the remainder of the article.
References
Abramson, C.J., Platt, S.R., Jakobs, C., Verhoeven, N.M., Dennis, R., Garosi, L. & Shelton, G.D., 2003, 'L-2-hydroxyglutaric aciduria in Staffordshire bull terriers', Journal of Veterinary Internal Medicine 17(4), 551-556. http://dx.doi.org/10.1111/j.1939-1676.2003.tb02477.x [ Links ]
Aghili, M., Zahedi, F. & Rafiee, E., 2009, 'Hydroxyglutaric aciduria and malignant brain tumor: A case report and literature review', Journal of Neurooncology 91(2), 233236. http://dx.doi.org/10.1007/s11060-008-9706-2 [ Links ]
Barth, P.G., Wanders, R.J., Scholte, H.R., Abeling, N., Jakobs, C., Schutgens, R.B. & Vreken, P., 1998, 'L-2-hydroxyglutaric aciduria and lactic acidosis', Journal of Inherited Metabolic Diseases 21(3), 251-4 http://dx.doi.org/10.1023/A:1005316121584 [ Links ]
Duran, M., Kamerling, J.P., Bakker, H.D., VanGennip, A.H. & Wadman, S.K., 1980, 'L-2-hydroxyglutaric aciduria: An inborn error of metabolism?', Journal of Inherited Metabolic Diseases 3(4), 109-112. http://dx.doi.org/10.1007/BF02312543 [ Links ]
Farias, F.H., Zeng, R., Johnson, G.S., Shelton, G.D., Paquette, D. & O'Brien, D.P., 2012, 'A L2HGDH initiator methionine codon mutation in a Yorkshire terrier with L-2-hydroxyglutaric aciduria', BMC Veterinary Research 8, 124. http://dx.doi.org/10.1186/1746-6148-8-124 [ Links ]
Garosi, L.S., Penderis, J., McConnell, J.F. & Jakobs, C., 2005, 'L-2-hydroxyglutaric aciduria in a West Highland white terrier', Veterinary Record 156(5), 145-147. http://dx.doi.org/10.1016/j.ymgme.2012.03.021 [ Links ]
Kolker, S., Boy, S.P., Heringer, J., Muller, E., Maier, E.M., Ensenauer, R., Muhlhausen, C., Schlune, A., Greenberg, C. R., Koeller, D.M., Hoffmann, G.F., Haege, G. & Burgard, P., 2012, 'Complementary dietary treatment using lysine-free, arginine-fortified amino acid supplements in glutaric aciduria type I - A decade of experience', Molecular Genetics and Metabolism 107(1-2), 72-80. http://dx.doi.org/10.1016/j.ymgme.2012.03.021 [ Links ]
Kranendijk, M., Struys, E.A., Salomons, G.S., Van der Knaap, M.S. & Jakobs, C., 2012, 'Progress in understanding 2-hydroxyglutaric acidurias', Journal of Inherited Metabolic Diseases 35(4), 571-587. http://dx.doi.org/10.1007/s10545-012-9462-5 [ Links ]
Patay, Z., Mills, J.C., Lobel, U., Lambert, A., Sablauer, A. & Ellison, D.W., 2012, 'Cerebral neoplasms in L-2 hydroxyglutaric aciduria: 3 new cases and meta-analysis of literature data', American Journal of Neuroradiology 33(5), 940-943. http://dx.doi.org/10.3174/ajnr.A2869 [ Links ]
Penderis, J., Calvin, J., Abramson, C., Jakobs, C., Pettitt, L., Binns, M.M., Verhoeven, N.M., O'Driscoll, E., Platt, S.R. & Mellersh, C.S., 2007, 'L-2-hydroxyglutaric aciduria: Characterisation of the molecular defect in a spontaneous canine model', Journal of Medical Genetetics 44(5), 334-340. http://dx.doi.org/10.1136/jmg.2006.042507 [ Links ]
Rzem, R., Veiga-da-Cunha, M., Noel, G., Goffette, S., Nassogne, M.C., Tabarki, B., Scholler, C., Marquardt, T., Vikkula, M. & Van Schaftingen, E., 2004, 'A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria', Proceedings of the National Academy of Sciences 101(48), 16849-16854. http://dx.doi.org/10.1073/pnas.0404840101 [ Links ]
Samuraki, M., Komai, K., Hasegawa, Y., Kimura, M., Yamaguchi, S., Terada, N. & Yamada, M., 2008, 'A successfully treated adult patient with L-2-hydroxyglutaric aciduria', Neurology 70(13), 1051-1052. http://dx.doi.org/10.1212/01.wnl.0000287141.90944.95 [ Links ]
Sanchez-Masian, D.F., Artuch, R., Mascort, J., Jakobs, C., Salomons, G., Zamora, A., Casado, M., Fernandez, M., Recio, A. & Lujan, A., 2012, 'L-2-hydroxyglutaric aciduria in two female Yorkshire terriers', Journal of the American Animal Hospital Association 48(5), 366-371. http://dx.doi.org/10.5326/JAAHA-MS-5967 [ Links ]
Scurrell, E., Davies, E., Baines, E., Cherubini, G.B., Platt, S., Blakemore, W., Williams, A. & Schoniger, S., 2008, 'Neuropathological findings in a Staffordshire bull terrier with l-2-hydroxyglutaric aciduria', Journal of Comparative Pathology 138(2-3), 160-164. http://dx.doi.org/10.1016/jjcpa.2007.11.005 [ Links ]
Short, A.D., Mellersh, C.S., Platt, H., Carter, S.D., Timofte, D., Lohi, H. & Ollier, W.E., 2010, 'Exonic mutations in the L2HGDH gene in Staffordshire bull terriers', Veterinary Record 167(12), 455-457. http://dx.doi.org/10.1136/vr.c4476 [ Links ]
Steenweg, M.E., Jakobs, C., Errami, A., Van Dooren, S.J., Adeva Bartolome, M.T., Aerssens, P., Augoustides-Savvapoulou, P., Baric, I., Baumann, M., Bonafe, L., Chabrol, B., Clarke, J.T., Clayton, P., Coker, M., Cooper, S., Falik-Zaccai, T., Gorman, M., Hahn, A., Hasanoglu, A., King, M. D., de Klerk, H. B., Korman, S. H., Lee, C., Meldgaard Lund, A., Mejaski-Bosnjak, V., Pascual-Castroviejo, I., Raadhyaksha, A., Rootwelt, T., Roubertie, A., Ruiz-Falco, M.L., Scalais, E., Schimmel, U., Seijo-Martinez, M., Suri, M., Sykut-Cegielska, J., Trefz, F.K., Uziel, G., Valayannopoulos, V., Vianey-Saban, C., Vlaho, S., Vodopiutz, J., Wajner, M., Walter, J., Walter-Derbort, C., Yapici, Z., Zafeiriou, D.I., Spreeuwenberg, M.D., Celli, J., den Dunnen, J.T., Vander Knaap, M.S. & Salomons, G.S., 2010, 'An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: A genotype-phenotype study', Human Mutation 31(4), 380-90. http://dx.doi.org/10.1002/humu.21197 [ Links ]
Van der Watt, G., Owen, E.P., Berman, P., Meldau, S., Watermeyer, N., Olpin, S.E., Manning, N.J., Baumgarten, I., Leisegang, F. & Henderson, H., 2010, 'Glutaric aciduria type 1 in South Africa- high incidence of glutaryl-CoA dehydrodenase deficiency in black South Africans', Molecular Genetics and Metabolism 10(2-3)1, 178-182. [ Links ]
Weimar, C., Schlamann, M., Krageloh-Mann, I. & Schóls, L., 2013, 'L-2 hydroxyglutaric aciduria as a rare cause of leukencephalopathy in adults', Clinical Neurology and Neurosurgery 115(6), 765-766. http://dx.doi.org/10.1016/jj.clineuro.2012.06.040 [ Links ]
Yilmaz, K., 2009, 'Riboflavin treatment in a case with l-2-hydroxyglutaric aciduria', European Journal of Paediatric Neurology 13(1), 57-60. http://dx.doi.org/10.1016/j.ejpn. [ Links ]
 Correspondence:
Correspondence:
Marlies Bóhm
21 King Edward Street, Newton Park
Port Elizabeth 6045
South Africa
Email: marlies@wol.co.za
Received: 15 May 2013
Accepted: 01 Nov. 2013
Published: 13 May 2014

