










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Chemistry
versão On-line ISSN 1996-840X
versão impressa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.78 Durban 2024
http://dx.doi.org/10.17159/0379-4350/2024/v78a08
RESEARCH ARTICLE
Evaluating the effect of structural reorientation on thermochemical properties of 1,4-diamino-3,6-dinitropyrazolo[4,3-c]pyrazole
Lamla Thungatha; Conrad Mahlase; Lisa Ngcebesha
Council for Scientific and Industrial Research (CSIR), Pretoria, South Africa
ABSTRACT
Isomers of 1,4-diamino-3,6-dinitropyrazolo[4,3-c]pyrazole (LLM-119) were designed by structural reorientation of the fused pyrazole rings and their respective substituents (-NO2 and -NH2). The structural reorientation involves structural rearrangement, which results in different structural isomers. Employing this approach, six structural isomers of LLM-119 were designed. The effect of structural reorientation (isomerisation and derivatives) on the enthalpy of formation, detonation properties, impact sensitivity, and density of these molecules is studied computationally. The computational methods used in this work yielded results that are close to the literature values for LLM-119, namely 519.54 kJ.mol-1 for enthalpy of formation, 1.80 g.ml-1 for density, 8359.3 m.s-1 for detonation velocity, and 31.0 Gpa for detonation pressure, with a relative error of 2% for enthalpy of formation, 2% for density, 0.05% for detonation velocity, and 4% for detonation pressure. The correlation of the structural reorientation to the calculated thermochemical and detonation properties of the molecules indicated that molecules with a -NO2 group attached to a carbon atom and -NH2 connected to a nitrogen atom maximise the enthalpy of formation and detonation velocity. The joining of pyrazole molecules has less effect on these parameters. The data shows that density and detonation pressure improved when both -NO2 or -NH2 functional groups were on the same side of the molecular structure. The structural reorientation gave rise to 3,4-dinitropyrazolo[3,4-c]pyrazole-1,6-diamine which exhibited optimal density and detonation performance compared to other molecules.
Keywords: LLM-119, Fused rings, Azole, structural isomers, detonation properties
INTRODUCTION
Energetic materials (EMs) are organic and inorganic molecules or formulations that store energy that gets released during deflagration or detonation up to initiation. EMs are classified as pyrotechnics, propellants, and explosives and are applied mainly in the aerospace, mining industry, and defence.1 The currently applied EMs have known shortcomings concerning energy output, sensitivity, and processibility.2 The gaps led to continuous research in finding better-performing EMs.3 The desired EMs should have higher detonation performance, lower sensitivity, and good thermal stability.4 However, high detonation performance and low sensitivity are not easy to achieve, but a good balance between these properties has been the focus of research.5 Most EMs are generally made of an organic scaffold with energetic functional groups as substituents, the organic skeleton acts as fuel (source of heat and gases), and energetic functional groups act as an oxidiser.6 Performance of EMs is known to be dependent on oxygen balance (composition), density, and heat of formation.7 The High Energy Density Materials (HEDMs) are of current attention due to their excellent performance these materials have higher energy release with a smaller charging volume, and most of them contain C, H, O, and N elements. The nitrogen content enhances the detonation properties of HEDMs, and this is due to the increased number of N-N, C=N, and N-C bonds of the molecule.8
Azole compounds are aromatic five-membered ring molecules that consist of a nitrogen atom and at least once non-carbon atom (nitrogen, oxygen, and sulfur). These nitrogen-containing compounds are known as a family of HEDMs, which comprises tetrazole, triazole, imidazole, and pyrazole. These differ in the number of nitrogen atoms in the ring.9 The literature shows that the nitrogen-containing azole compound meets most of the requirements when designing and synthesising new EMs.7,9 The nitrogen-based azole molecules can be made of single, coupled, and fused heterocyclic rings. The fused heterocyclic ring EMs are known as one of the primary focuses in research on EM.10-11
The 3,6-dinitropyrazolo[4,3-c]pyrazole molecule, also known as DNPP, consists of two fused Pyrazole rings and is substituted with a nitro group at positions 3 and 6 of the molecule. The DNPP was first made by Russian chemists and was determined to have excellent thermal stability with a decomposition temperature of 330-336 oC, low impact sensitivity of 15 J, friction sensitivity of 160 N, and density of 1.563 g.ml-1.10 The synthetic route for DNPP involves six steps, however, it gives a high yield and is scalable. The fused pyrazole units make this molecule predominantly planar, which is vital in achieving a higher packing density of EMs.12 DNPP is a precursor for 1,4-diamino-3,6-dinitropyrazolo[4,3-c]pyrazole (LLM-119). LLM-119 is synthesised by aminating DNPP at positions 1 and 4. This was obtained with better yield and high purity.12-13 LLM-119 has an exothermic peak at 253 oC, h50 value of 24 cm, and a density of 1.845 g.ml-1.14
LLM-119 is a fused pyrazole scaffold that can be prepared in higher yields and purity. Due to its planar backbone, it is plausible to give higher-density derivatives with only slight modification of the molecular energetic functional groups. This work explores the effect of structural isomerisation (structural reorientation) of LLM-119 by changing the positions of the energetic functional groups, repositioning the nitrogen atoms of the pyrazole rings, and keeping the molecular formula constant. The thermochemical and detonation properties of the molecules were calculated and compared. Enthalpies of formation were calculated using the Gaussian software and the G3 composite method. Detonation properties were estimated using the Kamlet-Jacobs equations. Enthalpy of sublimation, density, and impact sensitivity were obtained using empirical relationships based on the electrostatic potential.
COMPUTATIONAL METHODS
General
GaussView generated input files, and all the input structures were neutral. The calculations (i.e., optimisation and frequency) were done with the Gaussian 09 program, which is available within the CHPC cluster.15 Preliminary geometry optimisation of electronic structures of these molecules was done using density functional theory (DFT) with the B3LYP/6-311G (d,p) level theory without any symmetry restriction and with default convergence criteria.16-19 The harmonic vibrational frequency calculations of the optimised molecules were evaluated at the same level of theory as an optimisation to confirm that the stationary points obtained correspond to the true minima of the potential energy surface. Further calculations on the optimised structures were done using B3LYP/6-311++G (2df,2p) level theory for the calculations of enthalpy of sublimation. For calculations of molecular surface parameters that are required as input terms for density and impact sensitivity calculations, the B3LYP/6-31G(d,p) level theory and B3PW91/6-31G(d,p) level theory were used respectively to match what was used in the development of the electrostatic potential-based empirical relationships.20-22
Molecular structures
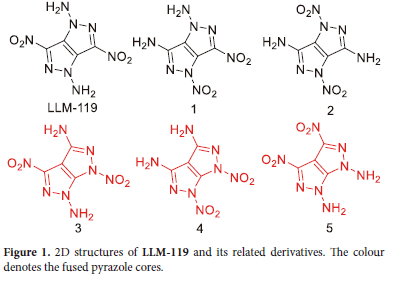
Molecular structures of 1,4-diamino-3,6-dinitropyrazolo[4,3-c] pyrazole (LLM-119, black in Figure 1) and 3,6-dinitropyrazolo[3,4-c] pyrazole-1,4(6H)-diamine (3,red in Figure 1) with their derivatives were generated for the computational calculations. The molecules used a similar pattern of substitution to obtain their respective derivatives. As these are structural isomers, these molecules have the same oxygen balance and nitrogen content.
Enthalpy calculations
The Gaussian compound method was utilised to obtain atomisation enthalpies of the molecules from the pre-optimised structures. The Gaussian-3 (G3) method is a quantum chemistry approach that uses ab initio molecular orbital calculations to estimate molecular energies of compounds with first and second-row atoms, evolving from G2 theory.23 The G3 method has been used to calculate enthalpies of formation for a set of energetic molecules with similar composition to that used here, resulting in a mean absolute deviation to experiment of 6 kcal mol-1, which was lower than the more computationally demanding G4 method.24 These molecules have the same molecular formula (C4H4N8O4), and their enthalpies of formation (0 K and 298 K) were obtained using the equations below:24
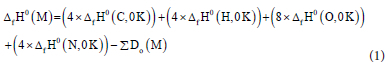
where ∆ is the gas-phase enthalpy of formation, ΣDo(M) is the atomisation enthalpy of the molecule. C, H, N, and O are carbon, hydrogen, nitrogen, and oxygen atoms.
The values for the enthalpies of formation for C, H, O and N atom which are 711.39, 216.03, 246.84 and 470.57 kJ-mol-1 respectively were obtained from the literature.25 The atomisation enthalpy of molecule M (C4H4N8O) can be obtained with the equation below.24, 26
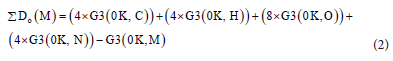
where G3 is the Gaussian composite method used to determine the enthalpy.
The enthalpy of formation at 298 K is obtained by the equations below.24,26 The values for G3 (0 K) and G3 (298 K) for the molecules are obtained directly from computational method G3.
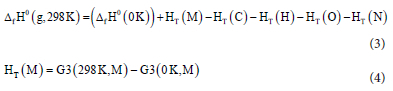
where M is (C4H4N8O4), C (carbon), H (hydrogen), O(oxygen), and N (nitrogen). HT (M) is the thermal correction for the molecule and HT (C), HT (H), HT (O), and HT (N) refer to thermal corrections for the elements in their standard states." The HT values for C, H, O and N were obtained from the literature with the values 1.05, 4.23, 4.34, 4.34 kJ-mol-1.27 The value for the molecule is obtained from the calculations.
The solid phase enthalpy of formation is determined as in Hess's law;28
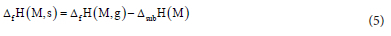
where ∆fH (M, g) is the gas-phase value and ∆subH (M) is the enthalpy of sublimation which can be calculated with equation 6;20

where A is the surface area of the molecule, v is the balance parameter for the positive and negative potential of the molecular surface and σtot2 represents total variance of the molecular overall surface potential.20 Here, the constants a, p, and y are 0.000267 kcal/mol/A4, 1.650087 kcal/mol, and 2.966078 kcal/mol respectively,29 The values of A, v, and σtot2 were calculated by optimising the molecules using the B3LYP/6-311++G (2df,2p) level theory and using Multiwfn as reported.17, 29-32
Detonation parameters
The performance of solid energetic materials is dependent on density. The density can be calculated using M/V0001, where M is the molecular mass of the molecule and Vm is the volume within 0.001 electrons Bohr-3 contour. The method ignores molecular interaction and as a result, it gives significant errors in some cases however, equation 7 accounts for these interactions;21 hence it is applied to determine the theoretical density of the molecules.

where a, p, and y are fitted constants which are 1.0462, 0.0021 g/(ml-kcal/mol2), and -0.1586 g.ml-1 respectively.33 The term v and σtot2 are as described in equation 6. The molecular structures were optimised using the B3LYP/6-31G(d,p) level theory,17,33 from which Vm, v, and σtot2 were calculated with Multiwfn.34
Detonation parameters (detonation velocity and detonation pressure) of CHNO energetic materials are calculated using Kamlet-Jacobs equations;35

where VD is the detonation velocity converted to m.s-1, and PD is the detonation pressure in GPa, N is the number of moles of gases per gram of explosive in mol.g-1, M is the average molecular mass of the gaseous products in g.mol-1, and Q refers to the chemical energy of detonation in kJ.g-1. The values of N, M and Q are estimated from the H2O-CO2 arbitrary decomposition assumption, and this require explosive's elemental composition and enthalpy of formation.35 Generally, for energetic molecules of category CaHbOcNd,4, 36-38, if , then equations 11-12 are applicable.
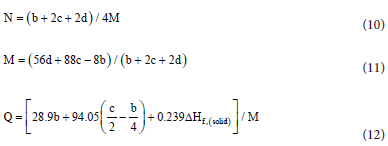
This criterion applies to the molecules studied here (C4H4N8O4). Therefore equations (10)-(12) are used to calculate N, M, and Q.
Impact sensitivity
Impact sensitivity, referred to as h50 (cm), is an important parameter. It gives information about how sensitive the energetic material is towards an external impact which is critical to operational safety. The lower the value of h50,the more sensitive the energetic molecule. This parameter can be measured experimentally using the drop weight method. The parameter can also be estimated theoretically, Pospisil et al.22 developed an equation to predict h50, which is based on molecular surface electrostatic potential.39

where a, β, and γ are fitted parameters and known to be -0.0064, 241.42, and -3.43, respectively.22 The term σ+2 is the variance of the positive surface potentials, and v is the degree of balance between a positive and negative potential of the molecular surface. The values σ+2 and v were obtained by optimising the molecules using the B3PW91/6-31G(d,p) level theory and using Multiwfn.18,22,32,34,40-41
RESULTS AND DISCUSSION
The molecular structure of LLM-119 has been used as a starting point for molecular design and modification to achieve related structural isomers. No additional substitution was done on the molecule. However, the way the pyrazole rings are fused was varied, and the substituents (-NO2 or -NH2) were either on the same side or on opposite sides of the molecule. As a result of these modifications, molecules 1-5 were achieved. The way pyrazole rings are fused for LLM-119, 1, and 2 is the same. Molecules 3-5 also share a similar fused pyrazole core structure which is different from LLM-119. Structural modification is the first step before computing and studying other properties. The optimised structures were taken as true minima with no imaginary frequencies.
Electronic structures
From the optimised structures, molecular electrostatic potential (MEP), highest occupied molecular orbitals (HOMO), and lowest unoccupied molecular orbitals (LUMO) were visualised. The MEP plot maps the distribution of electrostatic potential over the molecular isodensity surface, where the red-coloured area indicate negative regions, reactive to electrophiles, and the blue-coloured area are positive, reactive to nucleophiles.42 The MEP correlates with dipole moment, electronegativity, and partial charges,43 and makes it possible to identify reactive sites of a molecule toward nucleophiles and electrophiles. The MEP for the title compounds is shown in Figure 2. These were determined at 0.001 electrons per bohr3 isosurface. The electronic densities were determined at the B3LYP/6-311G(d,p) level of theory.44-45 For these molecules, the colour ranges from -0.06098 au (red) to 0.06098 au (blue), where the red area mainly lies over the -NO2 group and the blue over the -NH2 group. The positive regions are related to the impact sensitivity of the molecules.39
The HOMO and LUMO, also called frontier molecular orbitals, were also studied for the molecules to understand the electronic distribution and to analyse the HOMO-LUMO gaps; Figure 3 shows the 3D plot of these orbitals where the negative is shown in red and the positive in green. For all the molecules it can be observed that the HOMO electron density is less distributed at the -NO2 functional group while for LUMO the density is less at -NH2 groups. The HOMO, LUMO, and bandgap energies are also listed for each molecule (Table 1). The HOMO-LUMO gaps are known to be related to the kinetic stability and reactivity of the molecule.46 The more extensive the bandgap, the more stable and less reactive the molecule,47 molecules with -NO2 or -NH2 on the same side of the molecule have a higher bandgap (1, 4, and 5) This indicates the more excellent stability of these molecules.
Gas-phase and solid-phase enthalpy of formation
Here we investigate the derivatives of LLM-119 and its structural isomer to understand the effect of structural reorientation on the enthalpy of formation. The enthalpy of formation in EM is an important parameter as it is one of the inputs in determining other crucial performance parameters such as detonation pressure and detonation velocity. The higher and more positive this parameter is the higher its performance. The gas-phase enthalpies of formation are determined for all the molecules using the compound method (G3), and their values are listed in Table 1. The compounds exhibit positive enthalpy of formation. There are observed variations due to structural reorientation, with LLM-119 having the highest enthalpy, followed by molecule 5. The variation can be due to more than one factor. However, when -NO2 is attached via the carbon atom and -NH2 is attached via the nitrogen atom of the molecule, the energy is maximised. The fused pyrazole structural backbone seems to have an effect. LLM-119, 1 , and 2 structural backbones seem to give slightly higher enthalpies compared to 3, 4, and 5. The enthalpies of LLM-119, 1, and 2 structural backbones show significant variation due to substituents reorientation compared to molecule structural backbone of 3, 4, and 5.
Results also indicate that the trend of the total energies (in Hartrees), from the molecule with the highest energy to the one with the lowest energy, is LLM-119, 5, 3, 4, and 2. The higher the energy the molecule has, the lesser the thermodynamic stability. In this study, LLM-119 and molecule 5 are less stable.
From the gas-phase enthalpy of formation shown in Table 2, solid-phase enthalpies were calculated, as this is important for further calculations. Table 3 lists the calculated solid-phase enthalpy of formation for the molecules, the same trend is observed as in Table 1. The solid-phase enthalpy of formation for LLM-119 was compared with the literature value, which was calculated using the corrected Hartree-Fock method, and this gives a relative error of almost 2%, which justifies the reliability of the method used in this study.13 The relative error is calculated using the equation given in the support information.
Detonation properties and Impact sensitivity
Detonation properties are vital in determining the performance of EM; detonation velocity (D), detonation pressure (P) and heat of detonation (Q) are useful parameters in characterising EM. Fortunately, these can be obtained computationally with Kamlet-Jacobs empirical equations. The equation has been proven to be reliable in predicting detonation properties.48 In determining these parameters with Kamlet-Jacobs equations, Q and density (p) are required and the density is an essential factor in energetic materials. The molecules have constant oxygen balance and nitrogen content, which are -14% and 49.1%, respectively. These are important molecular makeup which contributes to the density and performance of EM. The oxygen balance indicates if the EM either has enough oxygen or less oxygen for the detonation reaction, oxygen-rich EM generates less toxic gas product. Table 4 lists the detonation parameters of the molecules and a comparison to the literature data.
From Table 4, LLM-119 has a calculated density of 1.80 g.ml-1 which slightly differs from the literature value with a relative error of 2%. The LLM-119 has the lowest density among the molecules, and 5 shows the highest. From the molecular structures, it seems that when the two -NO2 or -NH2 functional groups are on the same side of the molecule the density improves (1, 4 and 5). The fused pyrazole backbone of 3, 4 and 5 gives slightly higher density. The detonation velocities data for the molecules show that molecule 5 has the highest value of 8387.8 m/s which is somewhat higher than that of LLM-119, which is 8359.3 m/s. The detonation velocity for LLM-119 is close to the literature value with a relative error of 0.5%. Molecule 2 gives the lowest value, which is 8169.2 m/s. The detonation velocity of these molecules follows the trend from highest to lowest 5 > LLM-119 > 1 > 4 > 3 > 2. The structural properties show that the detonation velocity improves when the -NO2 group is attached to the carbon atom and when -NH2 is attached to a nitrogen atom of the molecule; hence molecule 5 and LLM-119 exhibit higher detonation velocity. The way the pyrazole molecules are fused has a slight effect on the detonation velocity. The detonation pressure data show that molecule 5 has the highest value of 31.6 GPa, and 2 shows the lowest value of 29.8 GPa. LLM-119 has a value of 31.0 GPa which is slightly higher than 1. The detonation pressure values from highest to lowest are 5 > 1 > LLM-119 > 4 > 3 > 2. The structural effect shows that the detonation pressure improves when the two -NO2 or -NH2 functional groups are on the same side of the molecule; this is the same as the trend observed with density. This is true since the detonation pressure is proportional to p2 as depicted by eq. 8. The LLM-119-based pyrazole fused core seems to give better detonation pressure than the fused pyrazole core of 3, 4 and 5. The relative error for LLM-119 detonation pressure is 4% which shows that the results are close to literature values.
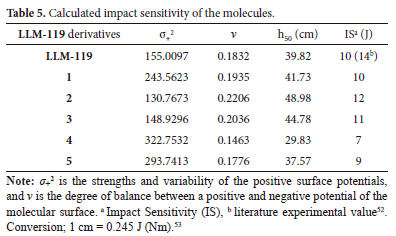
Impact sensitivity is another important parameter used to judge the sensitivity of EM to external impact; this parameter is designated as h50. The higher the h50 is, the more insensitive the explosive becomes. Table 5 lists the calculated impact sensitivity of the molecules. The comparison of calculated 10 J and the literature impact sensitivity of 14 J of LLM-119 gives a higher relative error of -29%, which means the calculated h50 is less than the one from the literature, h50 of 24 cm also appears in literature for LLM-119 which converts to be 6 J.51 Though our calculated h50 differ from both literature values it can be used to compare the molecules relatively. As can be seen from the data, the h50 increases from LLM-119 to 2 and then decreases to the lowest for molecule 4, which means it is the most insensitive, followed by 5 and LLM-119. Molecule 2 is sensitive compared to the rest of the molecules.
CONCLUSION
The structural reorientation of LLM-119 energetic material has been done, and six molecules were designed and optimised with DFT, and B3LYP (6-311 G(d,p) methods. All the calculations for the molecules were done successfully to obtain their gas-phase enthalpy, solid-phase enthalpies, detonation properties, impact sensitivity, density, and electronic properties. The structural reorientation has been correlated with the calculated thermochemical and detonation properties of the molecules. It was observed that molecules having -NO2 joined to a Carbon atom and -NH2 joined to the Nitrogen atom of the molecules maximise the enthalpy of formation and the detonation velocity. The way the pyrazole molecules are fused has a slight effect on the studied properties. The density and detonation pressure improve when both -NO2 or -NH2 functional groups are on the same side of the molecular structure. The structural reorientation gave rise to molecule 5, which is an isomer with optimal performance.
AUTHOR CONTRIBUTION
Dr. Lamla Thungatha and the late Dr. Xolani Peter identified the molecules to be studied. Dr Lamla did the molecular designs and calculation input files and wrote the work. Conrad Mahlase and Lisa Ngcebesha were the ones running the jobs obtaining the data from the computer cluster and doing further calculations for other parameters.
CONFLICTS OF INTEREST
The authors have no conflicts to declare.
ACKNOWLEDGEMENTS
This work was made possible by funding from the Armaments Corporation of South Africa (Armscor) supporting the Council of Scientific and Industrial Research (CSIR) research and development. The computation facility of the Centre for High-Performance Computing (CHPC) was utilised to conduct most of the molecular calculations via the computer cluster.
SUPPLEMENTARY MATERIAL
Supplementary data for this article can be found in the online supplement information.
ORCID ID
Lamla Thungatha: https://orcid.org/0000-0002-1104-5752
REFERENCES
1. Badgujar DM, Talawar MB, Asthana SN, Mahulikar PP. Advances in science and technology of modern energetic materials: an overview. J Hazard Mater. 2008;151(2-3):289-305. https://doi.org/10.1016/j.jhazmat.2007.10.039. [ Links ]
2. Sikder A, Sikder N. A review of advanced high performance, insensitive and thermally stable energetic materials emerging for military and space applications. J Hazard Mater. 2004;112(1-2):1-15. https://doi.org/10.1016/j.jhazmat.2004.04.003. [ Links ]
3. Zhang X, Zhu W, Xiao H. Theoretical studies on heats of formation, detonation properties, and bond dissociation energies of monofurazan derivatives. Int J Quantum Chem. 2010;110(8):1549-1558. https://doi.org/10.1002/qua.22283. [ Links ]
4. Lian P, Li Y, Li H, Huo H, Wang B, Lai W. A DFT study on the structure and property of novel nitroimidazole derivatives as high energy density materials. Comput Theor Chem. 2017;1118:39-44. https://doi.org/10.1016/j.comptc.2017.08.031. [ Links ]
5. Pan Y, Zhu W. Theoretical design on a series of novel bicyclic and cage nitramines as high energy density compounds. J Phys Chem A. 2017;121(47):9163-9171. https://doi.org/10.1021/acs.jpca.7b10462. [ Links ]
6. Fei T, Du Y, Pang S. Theoretical design and prediction of properties for dinitromethyl, fluorodinitromethyl, and (difluoroamino) dinitromethyl derivatives of triazole and tetrazole. RSC Adv. 2018;8(19):10215-10227. https://doi.org/10.1039/C8RA00699G. [ Links ]
7. Klapötke TM, Sabaté CM. Bistetrazoles: nitrogen-rich, high-performing, insensitive energetic compounds. Chem Mater. 2008;20(11):3629-3637. https://doi.org/10.1021/cm703657k. [ Links ]
8. Zhou H, Ma Z, Wang J, Wang D. Theoretical study of an energetic material di-1H-1, 3, 4-triazole derivatives. Defence Technology. 2014;10(4):384-392. https://doi.org/10.1016/j.dt.2014.08.002. [ Links ]
9. Gao H, Shreeve JM. Azole-based energetic salts. Chem Rev. 2011;111(11):7377-7436. https://doi.org/10.1021/cr200039c. [ Links ]
10. Thottempudi V, Yin P, Zhang J, Parrish DA, Shreeve JM. 1, 2, 3-Triazolo [4, 5,-e] furazano [3, 4,-b] pyrazine 6-Oxide - A Fused Heterocycle with a Roving Hydrogen Forms a New Class of Insensitive Energetic Materials. Chemistry. 2014;20(2):542-548. https://doi.org/10.1002/chem.201303469. [ Links ]
11. Thottempudi V, Forohor F, Parrish DA, Shreeve JM. Tris (triazolo) benzene and Its Derivatives: High-Density Energetic Materials. Angew Chem Int Ed. 2012;51(39):9881-9885. https://doi.org/10.1002/anie.201205134. [ Links ]
12. Pagoria PF, Lee GS, Mitchell AR, Schmidt RD. A review of energetic materials synthesis. Thermochim Acta. 2002;384(1-2):187-204. https://doi.org/10.1016/S0040-6031(01)00805-X. [ Links ]
13. Fried LE, Manaa MR, Pagoria PF, Simpson RL. Design and synthesis of energetic materials. Annu Rev Mater Res. 2001;31(1):291-321. https://doi.org/10.1146/annurev.matsci.31.1.291. [ Links ]
14. Zhang P, Kumar D, Zhang L, Shem-Tov D, Petrutik N, Chinnam AK, Yao C, Pang S, Gozin M. Energetic Butterfly: Heat-Resistant Diaminodinitro trans-Bimane. Molecules. 2019;24(23):4324. https://doi.org/10.3390/molecules24234324. [ Links ]
15. Frisch MJ, Clemente FR, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, et al. Gaussian 09, Revision A. 01. Wallingford (CT): Gaussian, Inc.; 2013. [ Links ]
16. Wang K, Shu Y, Liu N, Lai W, Yu T, Ding X, Wu Z. Theoretical studies on structure and performance of [1, 2, 5]-oxadiazolo-[3, 4-d]-pyridazine-based derivatives. J Phys Org Chem. 2017;30(1):e3591. https://doi.org/10.1002/poc.3591. [ Links ]
17. Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J Phys Chem. 1994 Nov;98(45):11623-11627. https://doi.org/10.1021/j100096a001. [ Links ]
18. Becke A. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J Chem Phys. 1993;98(7):5648-5652. https://doi.org/10.1063/1.464913. [ Links ]
19. Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter. 1988;37(2):785-789. https://doi.org/10.1103/PhysRevB.37.785. [ Links ]
20. Politzer P, Murray JS, Edward Grice M, Desalvo M, Miller E. Calculation of heats of sublimation and solid phase heats of formation. Mol Phys. 1997;91(5):923-928. https://doi.org/10.1080/002689797171030. [ Links ]
21. Politzer P, Martinez J, Murray JS, Concha MC, Toro-Labbé A. An electrostatic interaction correction for improved crystal density prediction. Mol Phys. 2009;107(19):2095-2101. https://doi.org/10.1080/00268970903156306. [ Links ]
22. Pospísil M, Vávra P, Concha MC, Murray JS, Politzer P. A possible crystal volume factor in the impact sensitivities of some energetic compounds. J Mol Model. 2010;16(5):895-901. https://doi.org/10.1007/s00894-009-0587-x. [ Links ]
23. Curtiss LA, Raghavachari K, Redfern PC, Rassolov V, Pople JA. Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J Chem Phys. 1998;109(18):7764-7776. https://doi.org/10.1063/L477422. [ Links ]
24. Manaa MR, Fried LE, Kuo I-FW. Determination of enthalpies of formation of energetic molecules with composite quantum chemical methods. Chem Phys Lett. 2016;648:31-35. https://doi.org/10.1016/j.cplett.2016.01.071. [ Links ]
25. Ruscic B. ATcT [1,2] enthalpies of formation based on version 1.130 of the Thermochemical Network [3]. https://atct.anl.gov/Thermochemical%20Data/version%201.130/index.php (accessed November 17, 2023). [ Links ]
26. Curtiss LA, Raghavachari K, Redfern PC, Pople JA. Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J Chem Phys. 1997;106(3):1063-1079. https://doi.org/10.1063/L473182. [ Links ]
27. Nicolaides A, Rauk A, Glukhovtsev MN, Radom L. Heats of formation from G2, G2 (MP2), and G2 (MP2, SVP) total energies. J Phys Chem. 1996;100(44):17460-17464. https://doi.org/10.1021/jp9613753. [ Links ]
28. Atkins PW. J. Phys. Chem. U. K.: Oxford University Press; 1982. [ Links ]
29. Byrd EF, Rice BM. Improved prediction of heats of formation of energetic materials using quantum mechanical calculations. J Phys Chem A. 2006;110(3):1005-1013. https://doi.org/10.1021/jp0536192. [ Links ]
30. Du M, Wang X, Guo Z. Theoretical design of bicyclo [2.2. 1] heptane derivatives for high-energy density compounds with low impact sensitivity. Comput Theor Chem. 2016;1095:54-64. https://doi.org/10.1016/j.comptc.2016.09.008. [ Links ]
31. Li X, Tang Z, Zhang X, Yang X. The heats of formation in a series of nitroester energetic compounds: A theoretical study. J Hazard Mater. 2009;165(1-3):372-378. https://doi.org/10.1016/j.jhazmat.2008.10.003. [ Links ]
32. Lu T, Chen F. Multiwfn: A multifunctional wavefunction analyzer. J Comput Chem. 2012;33(5):580-592. https://doi.org/10.1002/jcc.22885. [ Links ]
33. Rice BM, Byrd EF. Evaluation of electrostatic descriptors for predicting crystalline density. J Comput Chem. 2013;34(25):2146-2151. https://doi.org/10.1002/jcc.23369. [ Links ]
34. Lu T, Chen F. Multiwfn: A multifunctional wavefunction analyzer. J Comput Chem. 2012 Feb 15;33(5):580-592. https://doi.org/10.1002/jcc.22885. [ Links ]
35. Kamlet MJ, Jacobs S. Chemistry of detonations. I. A simple method for calculating detonation properties of C-H-N-O explosives. J Chem Phys. 1968;48(1):23-35. https://doi.org/10.1063/L1667908. [ Links ]
36. Tan B, Huang M, Long X, Li J, Fan G. Computational assessment of several hydrogen-free high energy compounds. J Mol Graph Model. 2016;63:85-90. https://doi.org/10.1016/j.jmgm.2015.11.002. [ Links ]
37. Qiu L, Xiao H, Gong X, Ju X, Zhu W. Theoretical studies on the structures, thermodynamic properties, detonation properties, and pyrolysis mechanisms of spiro nitramines. J Phys Chem A. 2006;110(10):3797-3807. https://doi.org/10.1021/jp054169g. [ Links ]
38. Qiu L, Gong X, Zheng J, Xiao H. Theoretical studies on polynitro-1, 3-bishomopentaprismanes as potential high energy density compounds. J Hazard Mater. 2009;166(2-3):931-938. https://doi.org/10.1016/j.jhazmat.2008.11.099. [ Links ]
39. Rice BM, Hare JJ. A quantum mechanical investigation of the relation between impact sensitivity and the charge distribution in energetic molecules. J Phys Chem A. 2002;106(9):1770-1783. https://doi.org/10.1021/jp012602q. [ Links ]
40. Durant JL. Evaluation of transition state properties by density functional theory. Chem Phys Lett. 1996;256(6):595-602. https://doi.org/10.1016/0009-2614(96)00478-2. [ Links ]
41. Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys Rev B Condens Matter. 1992;46(11):6671-6687. https://doi.org/10.1103/PhysRevB.46.6671. [ Links ]
42. Alphonse R, Varghese A, George L, Nizam A. Estimation of ground state and excited state dipole moments of a novel Schiff base derivative containing 1, 2, 4-triazole nucleus by solvatochromic method. J Mol Liq. 2016;215:387-395. https://doi.org/10.1016/j.molliq.2015.12.050. [ Links ]
43. Gandhimathi S, Balakrishnan C, Venkataraman R, Neelakantan MA. Crystal structure, solvatochromism and estimation of ground and excited state dipole moments of an allyl arm containing Schiff base: experimental and theoretical calculations. J Mol Liq. 2016;219:239-250. https://doi.org/10.1016/j.molliq.2016.02.097. [ Links ]
44. El Behi S, Ayachi S, Znaidia S. Computational modeling for the design of new fluorescent organic compounds based on both diketopyrrolopyrrole and nitrobenzofurazan moieties. J Mol Liq. 2022;360:119550. https://doi.org/10.1016/j.molliq.2022.119550. [ Links ]
45. Pathade VA, Waghchaure RH, Jagdale BS, Pawar TB, Pathade SS. Molecular structure, frontier molecular orbital and spectroscopic examination on dihydropyrimidinones: a comparative computational approach. Int J Adv Sci Res. 2020;11 Suppl 2:64-70. [ Links ]
46. Zeng Q, Qu Y, Li J, Huang H. A DFT study of five-membered nitrogen-containing fused heterocycles for insensitive highly energetic materials. RSC Adv. 2016;6(80):77005-77012. https://doi.org/10.1039/C6RA11624H. [ Links ]
47. Pan Y, Zhu W. Designing and looking for novel cage compounds based on bicyclo-HMX as high energy density compounds. RSC Adv. 2018;8(1):44-52. https://doi.org/10.1039/C7RA11584A. [ Links ]
48. Wei T, Zhu W, Zhang X, Li Y-F, Xiao H. Molecular Design of 1,2,4,5-Tetrazine-Based High-Energy Density Materials. J Phys Chem A. 2009;113(33):9404-9412. https://doi.org/10.1021/jp902295v. [ Links ]
49. Li Y, Shu Y, Wang B, Zhang S, Zhai L. Synthesis, structure and properties of neutral energetic materials based on N-functionalization of 3, 6-dinitropyrazolo [4, 3-c] pyrazole. RSC Adv. 2016;6(88):84760-84768. https://doi.org/10.1039/C6RA19556C. [ Links ]
50. Yin P, Zhang J, Mitchell LA, Parrish DA, Shreeve JM. 3, 6-Dinitropyrazolo [4, 3-c] pyrazole-Based Multipurpose Energetic Materials through Versatile N-Functionalization Strategies. Angew Chem Int Ed. 2016;55(41):12895-12897. https://doi.org/10.1002/anie.201606894. [ Links ]
51. Politzer P, Murray JS. Impact sensitivity and the maximum heat of detonation. J Mol Model. 2015;21(10):262. https://doi.org/10.1007/s00894-015-2793-z. [ Links ]
52. Li Y-N, Wang B-Z, Shu Y-J, Zhang S-Y, Lian P. Synthesis and Property of 1, 4-Diamino-3, 6-dinitropyrazolo [4, 3-c] pyrazole and Its Derivatives. Cent Eur J Energ Mater. 2016;13(2):321-333. https://doi.org/10.22211/cejem/64987. [ Links ]
53. Keshavarz MH, Klapötke TM. The properties of energetic materials: sensitivity, physical and thermodynamic properties. Walter de Gruyter GmbH & Co KG; 2021. https://doi.org/10.1515/9783110740158. [ Links ]
Received 12 June 2023
Revised 22 January 2024
Accepted 27 January 2024
* To whom correspondence should be addressed. Email: lthungatha@csir.co.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}