










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.77 Durban 2023
http://dx.doi.org/10.17159/0379-4350/2023/v77a21
RESEARCH ARTICLE
Synthetic Possibilities for Hemilabile Ligands: A Case Study of Decacyclo[10.8.15,8.02,11.04,9.013,20.015,18]-heneicosane-3,10,14,19-tetraone
Johan H. L. Jordaan; Frans J. Smit; Hermanus C. M. Vosloo; Agatha M. Viljoen
Research Focus Area for Chemical Resource Beneficiation: Catalysis and Synthesis Research Group, North-West University, Potchefstroom, South Africa
ABSTRACT
As proof of the synthetic possibilities for hemilabile ligands the chemistry of decacyclo[10.8.15,8.02,11.04,9.013,20.015,18]heneicosane-3,10,14,19-tetraone (5) was investigated. Reacting 5 with ethylene glycol under acid conditions gave the expected di-acetal protected ketone (6) as four possible isomers. Reduction of these isomers to produce the dialcohol ketal (7) was only possible with LiAlH4, after NaBH4, Luche's, and Meerwein-Ponndorf-Verley reduction methods were unsuccessful. Deprotection of 7 to the hydroxyl ketone (8) derivative was not possible under reflux with a 25% HCl solution. To evaluate the reactivity of 5, and investigate alternative synthetic routes to Grubbs pre-catalysis, 5 was treated with the reducing agents i) NaBH4, ii) glacial AcOH, Zn and iii) 80% AcOH/H2O/Zn mixture, which resulted in various reduction products. The AcOH/H2O/Zn reduction resulted in various products and a further investigation into the mechanism is given within this report.
Keywords: Pentacycloundecane, Grubbs pre-catalyst, Cyclo-addition, Hemilabile ligands
INTRODUCTION
As part of a program that is concerned with the design and synthesis of novel Grubbs-type pre-catalysts, containing the hemilabile pyridinyl alcholato ligand for the metathesis of 1-alkene derivates, these ligands convey increased thermal stability of the pre-catalysts and decreased the extent of side reactions.1-7 Incorporation of an alicyclic moiety in the hemilable ligands may further improve the thermal stability of these Grubbs-type pre-catalysts.8 Filipescu9 and Kusher10 were able to synthesize the Diels-Alder adduct (2) from the cyclo-addition of cyclopentadiene and 1,4-naphthoquinone (1) by intramolecular photocyclization to produce the expected hexacyclo[7.4.2.01,9.03,7.04,14.06,15]pentadeca-10,12-diene-2,8-dione (3, Scheme 1). Besides this compound's thermal stability,11 and thus the potential to bestow additional stability to the Grubbs-type pre-catalysts, it can also undergo further Diels-Alder reactions.12 Various researchers have shown that 3 can react exclusively either from the cyclobutane face or from the ketone face of the diene, depending on the nature of dienophiles (Figure 1).12-16 In 1987 Coxon et al.12 showed that due to the steric bulk of the p-benzoquinone, it reacts exclusively on the carbonyl face to produce the Diels-Alder adduct (4). If 4 is then irradiated with UV it undergoes a cyclization reaction to form the decacyclo-[10.8.15,8.02,11.04,9.013,20.015,18]heneicosane-3,10,14,19-tetraone (5, Scheme 1).10, 17, 18
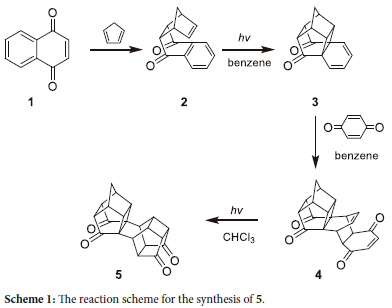
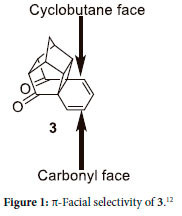
It was envisaged that cage compound 5, be used as a substrate for the synthesis of bidentate or monodentate hemilabile ligands for Grubbs-type pre-catalysts. However, before the hemilabile ligands are to be synthesized, an understanding of the chemistry and reactions of 5 is required. This paper will present the synthesis, characterization, and chemistry of derivatives of 5.
EXPERIMENTAL
The quantum-chemical calculations were carried out by density functional theory (DFT) since it usually gives realistic geometries, relative energies, and vibrational frequencies for transition metal compounds. All calculations were performed with the DMol3 DFT code as implemented in Accelrys Materials Studio* 4.2 using GGA/DNP/PW91 functional. The convergence criteria for these optimisations consisted of threshold values of 2 x 10-5 Ha, 0.004 Ha/A and 0.005 A for energy, gradient and displacement convergence, respectively, while a self-consistent field (SCF) density convergence threshold value of 1x10-5 Ha was specified. The electron density, frontier orbitais and Fukui-function was also calculated. Two computer systems were used for calculations, viz.
• HP Proliant CP4000 Linux Beowulf cluster, with 12 calculation nodes consisting of 4 HP DL145, 2 x 2.8 GHz AMD Opteron 64 CPU, 2 GB RAM, running Redhat Enterprise Linux 4
• HP Compaq dx2200 MT Intel® Core™2 Duo T7300 CPU @ 2.00 GHz, 3 GB RAM running Microsoft Windows XP Professional with service pack 2.
Experimental data were recorded using the following instruments: Infrared spectra (KBr discs) were recorded on a Bruker Tensor 27-IR spectrometer; EI mass spectra were obtained at 70 eV on a Micromass Autospec-TOF mass spectrometer and FAB mass spectra were obtained on a VG 70-70E magnetic sector analyser with matrix of nitrobenzyl alcohol (NBA). High-resolution MS spectra were obtained on a Bruker Micro-QTof II with an APCI source. NMR data were collected on a Varian Gemini-300 NMR spectrometer and a Bruker 600MHz Avance Ultrashield Plus. Melting points were determined using a Büchi Melting Point B-540 apparatus. Melting points are uncorrected.
Synthesis of 2:10
10 g (63.28 mmol) 1,4-naphthoquinone (1) was dissolved in 800 ml methanol, after which 10 g of activated carbon was added and stirred for 10 minutes on a hot plate. The solution was filtered to yield a yellow solution of 1,4-naphthoquinone. To this solution 2.5 g (6.86 mmol) cetyl trimethylammonium bromide (CTAB) was added, followed by the addition of 25 ml (297.28 mmol) freshly distilled cyclopentadiene. After 3 hours of stirring in the dark at room temperature, the solution was concentrated to a small volume and was poured into 500 ml water, upon which a white to light-peach coloured precipitate formed. The precipitate was filtered and washed with 10 ml cold water and 5 ml methanol under vacuum suction, successively. The crystals were dried to yield 12.03 g (53.69 mmol, 85 %) as a white powder. Melting point: 101 °C. IR (KBr): vmax 3443, 2992, 1680, 1589 and 1270 cm-1. MS (EI): M+ m/z 224. NMR: data were identical to the authentic samples.
Synthesis of hexacyclo[7.4.2.01,9.03,7.04,14.06,15]pentadeca-10,12-diene-2,8-dione (3):10
6 g (26.78 mmol) of the Diels-Alder adduct 2 was dissolved in 300 ml benzene and irradiated with a medium-pressure UV lamp for 2 hours in Pyrex vessels. After 2 hours the benzene was removed by reduced pressure which afforded an off-white solid, which was recrystallized from n-heptane to yield white-light yellow crystals (5.2 g, 23.21 mmol, 87%). Melting point: 109 °C. IR (KBr): vmax 3443, 2984, 1745, 1089 and 704 cm-1. MS (EI): M+ m/z 224. 13C-NMR [CDCl3, 150 MHz]: δC 210.33 (S, C=O), 124.67 (D, HC=CH), 119.73 (D, HC=CH), 54.538 (D), 51.57 (D), 50.10 (S), 44.16 (D), 38.91 (T, CH2) ppm. 1H NMR [CDCl3, 600MHz]: δH 5.96-5.90 (m, HC=CH), 5.38-5.31 (m, HC=CH), 3.31-3.0 (s), 2.98-2.93 (m), 2.77-2.76 (s), 1.98-1.93 (d) and 1.74-1.70 (d) ppm.
Synthesis of octacyclo[10.6.2.15,8.02,6.02,11.04,9.07,11.013,18]hene-icosa-15(19-diene-3,10(14,17-tetraone (4):12
4.8 g (21.42 mmol) of 3 was dissolved in 350 ml benzene to which 2.32 g (21.48 mmol) para-benzoquinone was added and refluxed for 20 h after which a yellow solid precipitated. The solid was filtered and washed with 40 ml, 50% water/methanol solution to remove any unreacted reactants. The yellow crystals were dried to yield 6.79 g (20,45 mmol, 95 %) of 4. Mp: 265°C, Lit. Mp: 265 - 267 °C. IR (KBr): vmax 2924, 1744, 1716, 1666, 1276 and 1062 cm-1. MS (EI): M+ m/z 332, HRMS (APCI): m/z calc. for C21H16O4 [M+]: 332.1043, found: 332.1056. 13C-NMR [CDCl3, 75 MHz]: δC 211.96 (S, C=O), 197.75 (S, C=O), 141.43 (D, HC=CH), 133.79 (D, HC=CH), 55.99 (D), 53.15 (S), 43.47 (D), 43.39 (D), 41.66 (D), 40.68 (T, CH2), 34.28 (D) ppm. 1H NMR [CDCl3, 300MHz]: δH 6.63 (s, HC=CH), 6.38-6.36 (m, HC=CH), 3.57-3.56 (s), 3.47-3.44 (m), 2.89-2.87 (t), 2.74-2.73 (d), 2.64-2.63 (d), 1.97-1.93 (d, CH2), 1.84-1.80 (d, CH2) ppm.
Synthesis of decacyclo[10.8.15,8.02,11.04,9.013,20.015,18]hene-icosane-3,10,14,19-tetraone (5):17
6 g (18.07 mmol) of 4 was dissolved in 600 ml chloroform and irradiated with a medium-pressure UV lamp for 3 hours in a Pyrex vessel. After 3 h a white solid precipitated which was filtered and washed with chloroform to remove any unreacted reagents (4.31 g, 12.98 mmol, 95 %). Mp: 371°C, Lit. Mp: >360°C. IR (KBr): vmax 2984, 1736, 1709, 1347, 1301, 1237, 1145 and 1095 cm-1. MS (EI): M+ m/z 332, HRMS (APCI): m/z calc. for C21H16O4 [M+]: 332.1043, found: 332.1083. 13C-NMR [CDCl3, 75 MHz]: δC 210.50 (S, C=O), 209.22 (S, C=O), 55.12 (D), 46.77 (S), 45.53 (D), 43.99 (D), 43.93 (D), 40.92 (D), 40.74 (T, CH2), 33.12 (D), 31.81 (D) ppm. 1H NMR [CDCl3, 300MHz]: δH 3.38-3.34 (m), 3.15-3.13 (m), 3.14-2.96 (m), 2.86-2.84 (s), 2.80-2.79 (s), 2.23-2.20 (m), 2.08-2.07 (d, CH2), 1.96-1.92 (d, CH2) ppm.
Synthesis of oxa-ketal (6):19, 20
5 g (15.6 mmol) of 5, 1.9 g (1.7 ml, 30.63 mmol) ethylene glycol and 0.3 g (1.74 mmol) _p-toluenesulfonic acid (PTSA) was added in a conical flask equipped with a magnetic stirrer and a Dean-Stark apparatus and refluxed in 300 ml toluene for 12 h. After this time, the toluene was removed under reduced pressure and a brown solid precipitated. The solid was washed with cold water and 10 ml cold methanol, to remove the unreacted ethylene glycol and PTSA, which liberated light brown-white crystals. The crystals were dried to yield 5.66 g (13.47 mmol, 89 %) of 6. Mp: 253 °C. IR (KBr): vmax 2970, 1743, 1332, 1111 and 942 cm-1. MS (EI): M+ m/z 420, HRMS (APCI): m/z calc. for C25H25O6 [M++H]: 421.1646, found: 421.1653. 13C-NMR [CDCl3, 150 MHz]: δC 214.04 (S, C=O), 213.94 (S, C=O), 213.41(S, C=O), 212.96 (S, C=O), 113.55 (S, O-C-O), 113.31 (S, O-C-O), 113.04 (S, O-C-O), 112.94 (S, O-C-O), 65.81 (T, O-CH2), 65.65 (T, O-CH2), 65.43 (T, O-CH2), 65.31 (T, O-CH2), 65.26 (T, O-CH2), 65.24 (T, O-CH2), 64.56 (T, O-CH2), 64.27 (T, O-CH2), 54.23 (D), 54.11 (D), 51.18 (D), 46.84 (S), 46.74 (S), 44.49 (D), 44.44 (D), 44.19 (D), 43.86 (D), 43.41 (D), 43.36 (D), 42.88 (D), 42.64 (D), 42.55 (D), 42.45 (D), 42.38 (D), 42.20 (D), 41.86 (D), 39.03 (T, CH2 of bridge), 38.98 (D), 38.19 (D), 35.59 (D), 35.24 (D), 32.79 (D), 32.04 (D), 31.47 (D), 31.10 (D), 30.33 (D) ppm.
Synthesis of the hydroxyl-ketal (7):
Method 1:
2 g of the diketal (6) was dissolved in 100 ml ethanol. The solution was cooled to 0 °C by means of an ice bath. 1.5 g sodium borohydride (NaBH4) was added in small amounts so that the temperature did not rise above 5 °C. After addition, the reaction was stirred for 2 h in the ice bath, after which it was quenched with the addition of a solution of 50 ml water and 5 ml HCl. The resulting mixture was subjected to rotary evaporation to remove the excess ethanol. Subsequently, the solution was extracted three times with 50 ml CH2Cl2. The combined organic layers were washed with a small amount of water and successively with brine. The organic layer was dried with MgSO4 and filtered, after which the solvent was removed by rotary evaporation. IR and MS analysis showed no trace of a hydroxyl group, but rather the diketal (6) as the only compound present.
Method 2 (Luche reaction):21,22
2 g of the diketal (6) was dissolved in a 0.4 M solution of CeCl3-7H2O in ethanol. The solution was cooled to 0 °C in an ice bath. 1.5 g sodium borohydride (NaB14) was added in small amounts so that the temperature did not rise above 5 °C. After addition, the reaction was stirred for 2 h in the ice bath, after which it was quenched with the addition of a solution of 50 ml water and 5 ml HCl. The resulting mixture was subjected to rotary evaporation to remove the excess ethanol. Subsequently, the solution was extracted three times with 50 ml CH2Cl2. The combined organic layers were washed with a small amount of water and successively with brine. The organic layer was dried with MgSO4 and filtered, after which the solvent was removed by rotary evaporation. IR and MS analysis showed no trace of a hydroxyl group, but rather the diketal (6) as the only compound present.
Method 3 (Meerwein-Ponndorf-Verley reduction):23
3 g of 6 and 5 g of isopropanol was added to 100 ml toluene to which 0.3 g Al(OiPr)3 was added. The reaction mixture was refluxed at 60 oC for 96 h. The solution was cooled and extracted with 3x75 ml CH2Cl2 and the combined organic layers were washed with 100 ml water. The organic layer was dried with MgSO4 and concentrated by means of rotary evaporation. IR and MS analysis showed no trace of a hydroxyl group, but rather the diketal (6) as the only compound present.
Method 4:20,24
1.2 g (31.62 mmol) LiAlH4 was added over a period of 30 minutes to a stirred solution of 4 g (9.52 mmol) of 7 in 100 ml dry THF. After addition, the solution was refluxed for 30 minutes and left to cool to room temperature. 200 ml H2O was added to decompose the reaction mixture. This solution was extracted with 3 x 50 ml dichloromethane, washed with water, dried over MgSO4 and the solution was condensed in vacuo. The brown oil that formed was dissolved in 2 ml dichloromethane and added to 20 ml petroleum ether. The milky solution was poured into a clean beaker and left to evaporate to yield 3.8 g (8.96 mmol, 94%) of 7 as a white powder. Mp: 223°C. IR (KBr): vmax 3427, 2966, 2890, 1468, 1454, 1320, 1282, 1269, 1150, 1102, 1068, 1027, 1003, 956 and 581 cm1. MS (EI): M+ m/z 424, HRMS (APCI): m/z calc. for C25H29O6 [M++H]: 425.1959, found: 425.1987. 13C-NMR [CDCl3, 150 MHz]: δC 115.70 (S, O-C-O), 115.69 (S, O-C-O), 115.12 (S, O-C-O), 115.07 (S, O-C-O), 75.67 (D), 73.79 (D), 65.76 (T, O-CH2-R), 65.60 (T, O-CH2-R), 65.08(T, O-CH2-R), 64.94 (T, O-CH2-R), 64.06 (T, O-CH2-R), 63.86 (T, O-CH2-R), 62.34 (T, O-CH2-R), 62.08(T, O-CH2-R), 47.70 (D), 47.63 (D), 47.46 (D), 47.29 (D), 44.58 (S), 43.89 (S), 43.71 (S), 43.63 (D), 43.47 (D), 43.16 (S), 41.89 (D), 41.83 (D), 41.26 (D), 41.07 (D), 40.96 (D), 40.57 (D), 40.37 (D), 39.80 (D), 39.61 (D), 39.31 (D), 38.25 (D), 38.22 (D), 38.00 (D), 36.93 (D), 35.57 (D), 35.49 (D), 34.89 (T, CH2 Bridge), 34.87 (T, CH2 Bridge), 34.74 (D), 34.62 (D), 32.70 (D), 31.38 (D) ppm. 1H NMR [CDCl3, 600MHz]: δH 6.13-6.00 (dd, OH); 5.07-5.01 (dd, OH).
Synthesis of hydroxy-ketone (8):25
1 g (2.36 mmol) of 7 was added to a stirring solution of a 24% HBr solution. This was refluxed for 12 hours after which the hot solution was poured over ice water. The solution was extracted with 3 x 50 ml CH2Cl2, washed with water, and dried over MgSO4. The CH2Cl2 was concentrated on a rotary evaporator to yield a small amount of clear oil. The oil was dissolved in 1 ml CH2Cl2 and poured into 20 ml petroleum ether. A white solid precipitated and was filtered off to yield 116 mg (0.35 mmol, 14%) of 8. IR (KBr): vmax 3423, 2964, 2867, 1720, 1333, 1304, 1276, 1150, 1131, 1076, 1056, 1004 and 923 cm-1. MS (EI): M+ m/z 336, HRMS (APCI): m/z calc. for C21H20O4 [M++H]: 336, found: 336. NMR data were inconclusive in the identification of the product, due to solubility problems.
Reaction of 5 with Zn/AcOH/H20:26
20 g (305.90 mmol) Zinc was activated with a small amount of hydrochloric acid (HCl), after which the Zn was washed with acetone and subsequently with water. The activated Zn was added to a stirred solution of 100 ml 80 % acetic acid/H2O (AcOH) and 1 g (3.01 mmol) of 5. This solution was refluxed for 5 h, after which it was subjected to rotary evaporation until only a small amount of acetic acid was left and a white solid precipitated. This solid was filtered off and water was added to the mother liquor after which 30 precipitated (96 mg). The remaining filtrate was dissolved in water and extracted with 3 x 50 ml CH2Cl2. The combined organic layers were washed with water and dried over MgSO4. The CH2Cl2 was concentrated on a rotary evaporator to yield 33 and 35 (558 mg) as a white solid. 30: Mp: 281°C, IR (KBr): vmax 3400, 2961, 2874, 1741, 1355, 1296, 1226, 1196, 1134, 1071, 1013, 951, 908, 864, 643 and 499 cm-1. MS (FAB): [M]+ m/z 352. 13C-NMR [DMSO, 150 MHz]: δC 215.13 (S, C=O), 109.54 (S, O-C-O), 108.10 (S, O-C-O), 84.12 (S), 55.32 (D), 49.85 (D), 49.71 (D), 49.66 (D), 49.52 (D), 47.89 (D), 47.72 (D), 47.63 (S), 45.44 (D), 44.76 (D), 43.58 (D), 38.92 (D), 38.57 (D), 37.96 (D), 37.05 (D), 36.75 (D), 31.72 (D) ppm. 1H NMR [DMSO, 300MHz]: δH 6.55 (s), 5.12 (s), 3.35 (s), 2.54-1.46 (a series of multiples), 1.48-1.46 (d, CH2 - Bridge), 1.381.36 (d, CH2 - Bridge) ppm. 31 and 32: Mp: 278°C IR (KBr): vmax 3307, 2969, 1740, 1332, 1273, 1228, 1156, 1066, 1013, 913, 850, 709 and 540 cm-1. MS (FAB): [M-H2O+H]+ m/z 337. 13C-NMR [DMSO, 150 MHz]: δC 215.10 (S, C=O), 215.04 (S, C=O), 116.06 (S, O-C-O), 114.69 (S, O-C-O), 84.25 (S), 84.20 (S), 77.71 (D), 76.36 (D), 55.31 (S), 55.25 (S), 49.88 (D), 49.86 (D), 49.70 (D), 49.66 (D), 48.11 (D), 47.93 (D), 47.91 (D), 47.78 (D), 47.70 (D), 47.66 (D), 45.50 (D), 45.42 (D), 45.25 (D), 45.08 (D), 44.74 (D), 44.70 (D), 42.77 (D), 41.45 (D), 40.73 (D), 40.04 (D), 38.74 (D), 38.67 (D), 38.56 (D), 38.53 (D), 38.03 (D), 37.53 (D), 37.48 (D), 37.03 (T, CH2 - Bridge), 37.01 (T, CH2 - Bridge), 35.52 (D), 32.54 (D), 30.33 (D) ppm. 1H NMR [DMSO, 300MHz]: δH 6.66 (s), 5.22 (s), 4.35-4.33 (t), 4.26-4.24 (t), 3.33 (s), 2.72 -2.71 (t), 2.67-2.66 (t), 2.61-2.56 (m), 2.52-2.40 (series of multiples), 2.36-2.34 (d), 2.26-2.22 (m), 2.14-1.88 (series of multiples), 1.75-1.72 (m), 1.481.46 (d, CH2-Bridge), 1.38-1.36 (d, CH2-Bridge) ppm.
Synthesis of the transannulated hydrate (25):
1g of 5 was added to a stirred solution of glacial acidic acid and zinc. After 24h of reflux, a white solid precipitated. The solution was cooled, filtered, and dried to yield 1.01 g of 25 as the exclusive product (96%). IR (KBr): vmax 3412, 2984, 1736, 1709, 1348, 1302, 1237, 1201, 1146, 1096, 911 and 575 cm-1. MS (FAB): [M+H]+: m/z 351. 13C-NMR [DMSO, 150 MHz]: δC 213.48 (S, C=O), 110.37 (S, O-C-O), 54.84 (D), 48.74 (S) 46.65 (D), 44.92 (D), 43.43 (D), 40.55 (T, CH2 - Bridge) 40.25 (D), 34.67 (D), 31.91 (D) ppm. 1H NMR [DMSO, 600MHz]: δH 6.82, 3.34, 2.93, 2.88, 2.74, 2.63, 2.49, 2.39, 2.20, 1.95, 1.92(d, 11.07 Hz), 1.80 (d, 10.84 Hz) ppm.
RESULTS AND DISCUSSION
It was found that for compounds 2-5b, all analytical data were in accordance with authentic samples. For 5, the melting point was higher than that published by Pandey et al.,17 however it was in the same order (374-375 °C) as that published by Tolstikov et al.18 The melting point of 371 °C was confirmed with thermogravimetric analysis (TGA). The discrepancy in melting points between the two authors could be ascribed to the limitation of the older apparatus used by Pandey. The mono addition of ethylene glycol to pentacycloundecane compounds was introduced by Eaton et al.19 in 1976. According to Eaton the steric bulk of the cage compound together with the introduction of the first acetal group hinders the introduction of a second acetal group on the adjacent carbonyl. Although this reaction is chemoselective it is not regioselective, resulting in the formation of four different isomers ( Figure 2). It can be observed from Figure 2 that 6a and 6c as well as 6b and 6d will result in only two isomers detected by NMR analysis. The 13C-NMR of 5 indicates that there are only two carbonyls present and only eleven signals are registered instead of the twenty-one carbons present, which designate that the adjacent carbonyls are equivalent. Thus any one of the carbonyls on each side can therefore form an acetal, but the formation of a second acetal on the same side is prevented, due to steric hindrance, as indicated by Eaton et al.19
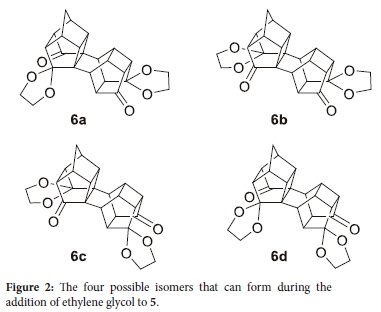
Contradictory to 5, the 13C-NMR of 6 shows almost double the number of peaks, 44 versus the expected 50 carbons for the expected isomers. The discrepancy in the number of peaks is due to some carbons having the same chemical shift (being degenerate) in both structures, for example, the DEPT shows only one CH2 bridge carbon and only two quaternary carbons, while there are two CH2 bridge carbons and four quaternary carbons respectively.27 Furthermore, the 13C-NMR of 6 shows four different carbonyl groups at δC 214.04, 213.94, 213.41 and 212.96 ppm. Figure 3 illustrates the numeric numbering of 6a and 6b used in Table 1, which shows the calculated C-C-O bond angles (Accelrys Materials Studio" 4.2 using GGA/DNP/PW91 functional). From this table, it was observed that although the angles do not differ significantly, this could be responsible for the slight chemical shift in the 13C-NMR. The distance between the oxygen of the acetal transannular to the carbonyl oxygen was also measured.
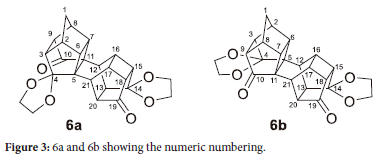
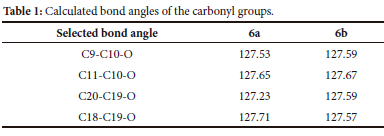
The carbonyl oxygen (on C10) and the acetal oxygen (on C3) distances are 3.26 Å and 3.23 Å for 6a and 6b, respectively, while the carbonyl oxygen (on C19) and the acetal oxygen (on C14) distance are 3.17 A and 3.15 A, respectively. The bond angles together with the slight difference in interatomic distances cause a minor change in the electronic environment around the carbonyls which causes the carbon atoms of 6a to be deshielded and shifted downfield, while the carbon atoms of 6b are more shielded and shifted upfield. This is commonly known as magnetic anisotropic systems and refers to the electron distribution of molecules with high electron density. This change in electron density affects the applied magnetic field on the different carbons and causes the observed chemical shift to change.28 This effect can be seen especially for the bridge CH2, where the two protons are split into two very distinct doublets in the 1H-NMR. Normally the carbonyl peak of C10 is downfield of the carbonyl peak of C19, which means that the chemical shift at δC 214.04 ppm, can be assigned to C10 in 6a, 213.94 ppm can be assigned to C10 in 6b, likewise, 213.41 ppm can be assigned to C19 in 6a, and 212.96 ppm can be assigned to C19 in 6b.
Since two distinguishable isomers are forming during the addition of ethylene glycol to 5 and conventional separation techniques, such as column chromatography, are unable to separate/purify these cage compounds, all the following products will have two or more isomers. Thus, for simplicity, the numeric value alone will imply all the isomers of a specific compound, while the alphabetical letter together with the numeric value will imply the specific isomer.
For the reduction of 6 to 7 the first method employed was the reduction of a carbonyl with NaBH4 (Scheme 2). Since the carbonyls of 6 are sterically blocked on one side by the acetal group, only exo hydride attack can occur, producing only the endo product. However, this method was unable to reduce the carbonyls of 6 and only the starting compound could be extracted in high yields.
Since NaBH4 alone were incapable of reducing the carbonyl groups of 6, the Luche21, 22 reaction was attempted. The Luche reaction is a selective reduction method for the reduction of enones or ketones in the presence of aldehydes. The activity of the Luche reaction can be explained by the Hard and Soft Acid and Base (HSAB) theory. Carbonyl groups require hard nucleophiles for the addition of a nucleophile to the carbonyl. The hardness of the borohydride is increased by replacing the hydride groups with alkoxide groups. This reaction is catalysed by cerium salts by increasing the electrophilicity of the carbonyl groups.22 Thus, a stronger reducing agent is produced and the cerium facilitates the reduction by binding to the oxygen, thereby weakening the carbonyl bond and making it more susceptible to reduction. Even with the ketone being more susceptible to reduction no reaction took place.
As a last option, LiAlH4 was used as a reducing agent.20, 24 It was possible to reduce 6 to 7, with relative ease and high yields (94%). This reaction was initially conducted with LiAlH4 dissolved in THF and the ketone being added portion-wise, over a period of 2 hours. It was argued that since the ketone is resistant to reduction this setup will result in a higher concentration of H- (hydride) at any given time compared to the concentration of the ketone. The resulting product was an orange-brown oil that we were unable to crystalize, however, the IR showed a distinctive OH stretching band at 3427 cm-1 and the disappearance of the carbonyl peak at 1743 cm-1, however, the resultant reaction mixture was highly contaminated, and we were unable to purify this any further. The reaction was therefore repeated in the same manner as before, except that LiAlH4 was added slowly to the stirred ketone (6) in THF over a period of 2 hours which resulted in 7 as an off-white powder. 13C NMR confirmed the disappearance of the carbonyl peaks. As with 6, the number of signals attributed to the different inseparable isomers made full elucidation problematic, however, the HRMS indicated that the calculated and measured m/z were in accordance to compound 7 (calc. for C25H29O6 [M++H]: 425.1959, found: 425.1987).
As with 6, there are two distinguishable compounds giving rise to eight different CH2-O-R carbon peaks between δC 65 and 62 ppm. The DEPT indicate two CH2 bridge peaks that were degenerate in 6. In comparison to 6, 7 has four different quaternary carbons, indicating that the isomer effect is more prominent in 6 than in 7. This can be explained by taking into consideration that the hydroxyl group is spatially larger than the ketone group, which has an increased electronic effect on the C3, C10, C14 and C19 of the two isomers.
The logical next step would then be the removal of the protecting acetal group of 7 to produce the hydroxyl ketone 8. For the pentacyl-coundecane system, this can normally be accomplished by stirring the ketol 7 in a 10% HCl solution for 2 hours under reflux. For 7 this procedure was inadequate to remove the acetal group. Even when the solution was refluxed for 6 days in a 25% HCl solution no reaction took place.
To test the reactivity of the 4 carbonyl groups of 5, it was subjected to reduction with NaBH4. The expected product was a tetraol, 15 (Scheme 3), but the triol 16a and 16b, as the major products with 17 as a minor product were obtained.
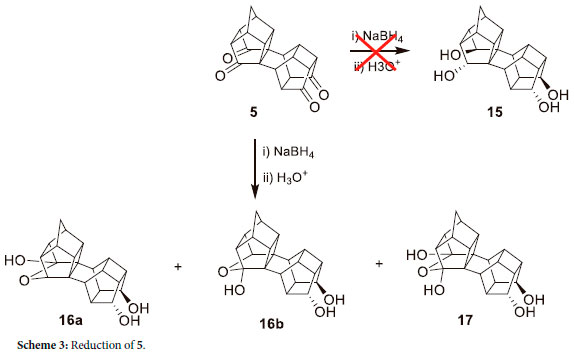
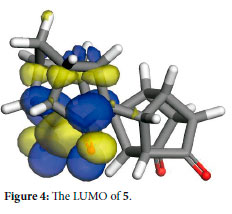
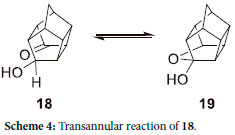
Molecular modelling indicates that the LUMO of 5 (Figure 4) is concentrated on the CH2-bridge side of the molecule and thus it is expected to be reduced first. It has been reported by Mehta et al.29 that for the pentacycloundecane system, the hemiacetal 18 is only observed in equilibrium with the ketol 19 (Scheme 4). It should be noted that the two carbonyl groups are equivalent and the reaction occurs without regiospecificity, although it was pointed out by Sasaki et al.30 that 18 did not cyclise to 19, even at 270 °C. Whether or not this cyclization occurs, it is evident from the 13C-NMR that only 16a and 16b are present, with 17 as the minor product, in this case.
Although physical evidence such as δC at 78.80 and 78.65 ppm are indications of a transannular ether-bearing carbon with hydrogen (instead of the normal hydroxyl group attached to this carbon). DEPT135 indicates that there are five quaternary carbons at δc 44.99, 44.51, 44.28, 43.90 and 43.80 ppm, four originating from 16a, 16b and one for 17 (since 17 is symmetrical). From the reaction in Scheme 3, it seems that all four ketones are active towards reduction. However, due to the steric hindrance around the ketones in 6, conveyed by the steric bulk of the acetal groups, they may be resistant to reduction.
In 1981 Mehta et al.26 showed that 20 can be directly synthesized from 3 with the use of zinc and acetic acid. It was argued that this reaction will allow the direct synthesis of 21, a hydroxyl ketone with the correct functionality, akin to 8 (Scheme 5).
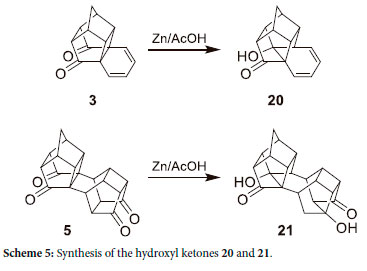
Initially, the reaction was carried out with the use of 99.9% acetic acid. However, 13C-NMR elucidation indicated a symmetrical product with the presence of only one carbonyl as well as a hydrate (O-C-O) group at δC 213.47 and 110.37 ppm, respectively, which correlate s to the NMR data of 25. Scheme 6 shows a possible reaction mechanism for the hydrate 25 formation as well as the formation of the hydroxyl ketone (30). It was posited that for the reaction to occur a hydrate must first form and thus there is insufficient water to solvate the acetic acid to release a proton that initiates the hydroxyl ketone formation (30). For this reason, the reaction stops at 25. This is like the reduction of 5 with NaBH4 (Scheme 3), of which 25 is a minor product that is reduced to 17 during the reaction. 13C-NMR of 25 showed only one carbonyl group, indicating that the transannular hydrate formation is chemoselective and consequently forms only one symmetrical product (an interesting observation from the 1H-NMR of this compound is the number of singlet peaks). This can be ascribed to the fact that within some cage compounds, 1, 2 and 3 bond coupling can be observed which results in extremely small coupling constants (J) which consequently results in the multiplet (that should have formed) being displayed as singlets.28 Furthermore, it depicts that when the first hydrate forms on one side, the reaction stops, and the addition of a second hydrate does not occur. It also sheds some light on the mechanism of the reaction, i.e., non-concerted manner.
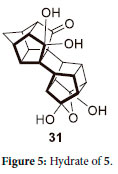
Galin et al.31 showed that it is possible to hydrate 5 to form 31 by reacting 5 in an aqueous acetone solution for three days (Figure 5). The findings of Galin are supportive of the proposed hydration mechanism as shown in Scheme 6.
Repeating the reaction with an 80% acetic acid solution for 5 hours, the 13C-NMR showed a mixture of products which included three hydroxyl-bearing carbons at δC 84.12, 84.25 and 84.20 ppm, which is consistent with a tertiary alcohol.28 The 13C-NMR also shows three different carbonyl groups at δC 215.13, 215.10 and 215.04 ppm. The spectrum also indicated that there still was a hydrate present together with two geminal alcohol groups. These products were separated by means of their differences in solubility. 13C-NMR of the product removed by filtration and washed with water (30), indicated only one carbonyl peak at δC 215.13 as well as the presence of a hydrate (O-C-O) at δC 109.5 ppm.
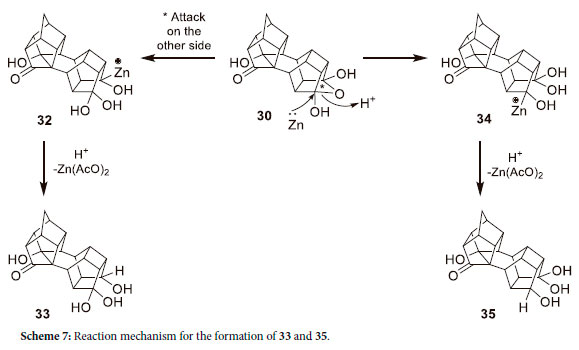
The 13C-NMR of the other fraction isolated (33 and 35), signifying that there were 42 resonance signals of which there were two carbonyl peaks at δC 215.1 and 215.04, two O-C-O peaks at δC 116.06 and 114.69, two different tertiary hydroxyl bearing carbon atoms at δC 84.25 and 84.20, two secondary hydroxyl bearing carbon atoms at δC 77.71 and 76.36, as well as two quaternary carbons at δC 55.31 and 55.25 ppm. This indicated that there were two isomers of which the degenerate peaks were split due to the isomer effect. Furthermore, the product contained a geminal alcohol together with an additional hydroxyl group. Scheme 7 shows the possible reaction mechanism for the formation of 33 and 35.
From these results it seemed that the formation of 25 is chemoselective and that only one hydrate is formed during the reduction of 5 with Zn/AcOH, resulting in only one side forming a hydroxyl ketone.
When the Zn/AcOH reduction reaction of 5 was carried out for 24 hours, instead of the original 5 hours with 80% AcOH, 33 and 35 are produced exclusively, confirming that 30 is a precursor for these compounds. It seems that the Zn/AcOH reduction of 5 does not form the double transannular hydroxy ketone 21, but rather the reaction proceeds through the formation of 25 and subsequent reduction of 25 to form 30, which reacts further with zinc in acetic acid to produce 33 and 35.
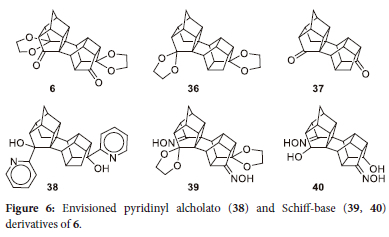
Although these are interesting results as to the derivatization of 5 showing the possibilities, it would be advantageous to be able to synthesize the Schiff-base (40) or the pyridinyl alcholato (38) derivatives of 5 (Figure 6). For these can act as ligands for the Grubbs catalysts. Attempts to derivatise 6 to 38 via 36 or 37 by means of the Huang-Minlon reaction were unsuccessful. Similarly, were the attempts to react 6 with hydroxyl amine to produce 39 unsuccessful. In all these cases only the starting material 6 was recovered.
CONCLUSION
In the process of developing novel hemilabile ligands, it was important to understand the chemical and physical properties of the tetraone 5. It seems that 5 shows similar chemistry as 3 when reacted with ethylene glycol to produce the corresponding acetal isomers 6a to 6d. Paradoxically, the isomers of 6 did not react with, Luche's or Meerwein-Ponndorf-Verley's reagents to give the corresponding hydroxyl acetal derivative 7, as it would have with 3. It seems that stronger reducing agents were to be used to convert 6 to 7. From the results obtained with the zinc/acetic acid reduction, it became clear that the chemistry of 5 is different from that of 3. With two active reducing sites present, the ketones on the CH2-bridge side were the first to undergo chemoselective hydration through a transannular mechanism. Following the formation of 25 it is further hydrated to form 30 and ultimately to 33 and 35. This is not surprising, since the transannular distance between the carbonyl groups is shorter on the CH2-bridge and the pi-orbitals show an overlap with molecular modelling. What is interesting though is that the extent of hydration can be controlled. Therefore, the chemistry which applies to 3 is not always applicable to that of 5. This was seen with the deprotection of the ketone groups in 7. It was also found that some of these compounds became less soluble and ultimately further reactions were not possible. This presents a problem with the synthesis of the possible ligands 38-40. With the knowledge gained from this investigation, it should be possible to devise a synthetic strategy to synthesise these ligands.
ACKNOWLEDGEMENTS
The authors would like to thank Prof Cornie van Sittert, for her help with the molecular modelling. Andre Joubert, for collection of NMR data. Prof. F.J.C. Martins for his assistance in the NMR data assignment. The National Research Foundation, c*Change and the North-West University for financial support.
SUPPLEMENTARY MATERIAL
Supplementary information for this article is provided in the online supplement.
ORCID IDS
Johan H. L. Jordaan: https://orcid.org/0000-0002-8134-6753
Frans J. Smit: https://orcid.org/0000-0003-1956-6584
Hermanus C. M. Vosloo: https://orcid.org/0000-0002-5879-323X
REFERENCES
1. du Toit JI, Jordaan M, Huijsmans CA, Jordaan JH, van Sittert CG, Vosloo HC. Improved metathesis lifetime: chelating pyridinyl-alcoholato ligands in the second generation grubbs precatalyst. Molecules. 2014;19(5):5522-5537. https://doi.org/10.3390/molecules19055522. [ Links ]
2. du Toit JI, van der Gryp P, Loock MM, Tole TT, Marx S, Jordaan JHL, Vosloo HCM. Industrial viability of homogeneous olefin metathesis: beneficiation of linear alpha olefins with the diphenyl-substituted pyridinyl alcoholato ruthenium carbene precatalyst. Catal Today. 2016;275:191-200. https://doi.org/10.1016/jxattod.2015.12.004. [ Links ]
3. Tole TT, Jordaan JHL, Vosloo HCM. A-pyridinyl alcohols, a,a'-pyridine diols, a-bipyridinyl alcohols, and a,a'-bipyridine diols as structure motifs towards important organic molecules and transition metal complexes. Curr Org Synth. 2020;17(5):344-366. https://doi.org/10.2174/1570179417666200212111049. [ Links ]
4. Tole T, Jordaan J, Vosloo H. Synthesis and application of the transition metal complexes of a-pyridinyl alcohols, a-bipyridinyl alcohols, a,a'-pyridinyl diols and a,a'-bipyridiny diols in homogeneous catalysis. Molecules. 2018;23(4):896-953. https://doi.org/10.3390/molecules23040896. [ Links ]
5. Tole TT, du Toit JI, van Sittert CGCE, Jordaan JHL, Vosloo HCM. Synthesis and application of novel ruthenium catalysts for high temperature alkene metathesis. Catalysts. 2017;7(12):22-43. https://doi.org/10.3390/catal7010022. [ Links ]
6. Tole TT, Jordaan JHL, Vosloo HCM. Synthetic methods of a-pyridinyl and a-bipyridinyl alcohols and a,a'-pyridine and a,a'-bipyridine diols. Curr Org Synth. 2015;12:261-304. https://doi.org/10.2174/1570179412666150218201902. [ Links ]
7. Tole TT, Jordaan JHL, Vosloo HCM. Catalysis of linear alkene metathesis by grubbs-type ruthenium alkylidene complexes containing hemilabile a,a'-diphenyl-(monosubstituted-pyridin-2-yl)methanolato ligands. Beilstein J Org Chem. 2019;15:194-209. https://doi.org/10.3762/bjoc.15.19. [ Links ]
8. Dinger MB, Nieczypor P, Mol JC. Adamantyl-substituted n-heterocyclic carbene ligands in second-generation grubbs-type metathesis catalysts. Organometallics. 2003;22(25):5291-5296. https://doi.org/10.1021/om034062k. [ Links ]
9. Filipescu N, Menter JM. Cage formation subsequent to intramolecular triplet-energy transfer. J Chem Soc B. 1969;616-620:616. https://doi.org/10.1039/j29690000616. [ Links ]
10. Kushner AS. Photoisomerization of the 1,4-naphthoquinonecyclopentadiene adduct. A novel intramolecular [6 + 2] cycloaddition. Tetrahedron Lett. 1971;12(35):3275-3278. https://doi.org/10.1016/S0040-4039(01)97154-0. [ Links ]
11. MehtaG, SinghV,RaoKS. Onthefluxionalbehaviourofapolycyclic(4.4.2) propella-2,4-diene. Tetrahedron Lett. 1980;21(14):1369-1372. https://doi.org/10.1016/S0040-4039(00)74579-5. [ Links ]
12. Coxon JM, O'Connell MJ, Steel PJ. Pi-facial selectivity in diels-alder reactions of hexacyclo[10.2.1.02,11.04,9.04,14.09,13]pentadeca-5,7-diene-3,10-dione. J Org Chem. 1987;52(21):4726-4732. https://doi.org/10.1021/jo00230a014. [ Links ]
13. Pandey B, Rao AT, Dalvi PV, Kumar P. Studies on cyclobutyl bond cleavage by adjacent ketyl radical generated under pet conditions. Tetrahedron. 1994;50(12):3835-3842. https://doi.org/10.1016/S0040-4020(01)90403-X. [ Links ]
14. Marchand AP, Zope UR, Burritt A, Bott SG, Coxon JM. A facially dissymmetric 1,3-cyclohexadiene as a dienophile in diels-alder reactions with polyhalocyclopentadienes. Tetrahedron. 1995;51(34):9319-9326. https://doi.org/10.1016/0040-4020(95)00519-E. [ Links ]
15. Coxon JM, Fong ST, Lundie K, McDonald Q, Steel PJ, Marchand AP, Zaragoza F, Zope UR, Rajagopal D, Bott SG, et al. Diastereofacial selectivity in diels-alder cycloadditions of methyl acrylate to facially differentiated unsymmetrical cyclohexa-1,3-dienes. Tetrahedron. 1994;50(45):13037-13048. https://doi.org/10.1016/S0040-4020(01)81221-7. [ Links ]
16. Mehta G, Uma R; Remarkable Stereodirecting Influence of Distal Protective Groups. Role of heteroatoms in diastereofacial control in cycloaddition to a dissymmetric cyclohexa-1,3-diene moiety in a polycyclic framework. Remarkable stereodirecting influence of distal protective groups. J Org Chem. 2000;65(6):1685-1696. https://doi.org/10.1021/jo991349b. [ Links ]
17. Pandey B, Zope UR, Ayyanger NR. Pi-facial stereoselectivity in diels-alder reaction of hexacyclo [7.4.2.01,9.03,7.04,14.06,15] pentadecane-10,12-diene-2,8-dione. Synth Commun. 1989;19:585-596. https://doi.org/10.1080/00397918908050703. [ Links ]
18. Tolstikov GA, Lerman BM, Galin FZ. Synthese eines neuen dekacyclo-heneikosans. Zh Org Khim. 1975;11:1348-1349. https://doi.org/10.1002/chin.197538215. [ Links ]
19. Eaton PE, Cassar L, Hudson RA, Hwang DR. Synthesis of homopentapris-mane and homohypostrophene and some comments on the mechanism of metal ion catalyzed rearrangements of polycyclic compounds. J Org Chem. 1976;41(8):1445-1448. https://doi.org/10.1021/jo00870a034. [ Links ]
20. Dekker TG, Oliver DW. Synthesis of (d3)-trishomocuban-4-ol via carbenium ion rearrangement of pentacyclo [5.4. 0.02, 6.03, 10.05, 9] undecan-8-ol. S Afr J Chem. 1979;32:45-48. https://doi.org/10.10520/AJA03794350_734. [ Links ]
21. Marchand AP, LaRoe WD, Sharma GVM, Suri SC, Reddy SD. Facile stereoselective reductions of enediones and cage diketones using sodium borohydride-cerium(III) chloride. J Org Chem. 1986;51(9):1622-1625. https://doi.org/10.1021/jo00359a054. [ Links ]
22. Luche JL. Lanthanides in organic chemistry. 1. Selective 1,2 reductions of conjugated ketones. J Am Chem Soc. 1978;100(7):2226-2227. https://doi.org/10.1021/ja00475a040. [ Links ]
23. Yin J, Huffman MA, Conrad KM, Armstrong JD. Highly diastereoselective catalytic meerwein-ponndorf-verley reductions. J Org Chem. 2006;71(2): 840-843. https://doi.org/10.1021/jo052121t. [ Links ]
24. Martinez GM, Vilar ET, Fraile A, de la Moya Cerero S, Ruiz PM. Synthesis of enantiopure norbornane derivatives. Effect of bridgehead substituent on the n-facial selectivity of the reduction of 2-norbornanones and their oximes. Tetrahedron Asymmetry. 1998;9(10):1737-1745. https://doi.org/10.1016/S0957-4166(98)00132-3. [ Links ]
25. Martins FJC, Viljoen AM, Kruger HG, Joubert JA. Synthesis of 8,11-dihydroxy-pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-lactam. Tetrahedron. 1993;49(42):9573-9580. https://doi.org/10.1016/S0040-4020(01)80226-X. [ Links ]
26. Mehta G, Rao KS, Bhadbhade MM, Venkatesan K. Novel rearrangement to a pentacyclopentanoid (polyquinane) system. J Chem Soc Chem Commun. 1981;755-756(15):755. https://doi.org/10.1039/c39810000755. [ Links ]
27. Kelani MT, Dieudonné D, Skowron KJ, Pedigo C, Govender T, Kruger HG, Naicker T, Onajole OK. Trends in nmr structural elucidation of polycyclic cages, namely: Adamantane, pentacycloundecane and trishomocubane. S Afr J Chem. 2021;75:117-129. https://doi.org/10.17159/0379-4350/2021/v75a14. [ Links ]
28. Silverstein RM (ed.). Spectrometric identification of organic compounds, 8th edition. Hoboken: Wiley, 2015. [ Links ]
29. Mehta G, Singh H, Pandey PN, Chaudhury B. Revelation of pentacyclo (6.2.1.02,7.04,10.05,9) undecane/oxa-bird cage equilibrium by 13CMR spectroscopy: fragmentation of caged polycyclic systems with lead tetraacetate. Chem Lett. 1980;9(1)59-62. https://doi.org/10.1246/cl.1980.59. [ Links ]
30. Sasaki T, Eguchi S, Kiriyama T, Hiroaki O. Studies on hetero-cage compounds - VI. Tetrahedron 1974;30:2707-2712. https://doi.org/10.1016/S0040-4020(01)97433-2. [ Links ]
31. Galin FZ, Afonichev DD, Lerman BM, Kazakov VP, Tolstikov GA. Zh Org Khim. 1978;14:2308-2317. [ Links ]
Received 19 April 2023
Revised 4 August 2023
Accepted 27 September 2023
* To whom correspondence should be addressed. Email: johan.jordaan@nwu.ac.za or frans.smit@nwu.ac.za


{kind=link}
{kind=link}