










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Chemistry
versão On-line ISSN 1996-840X
versão impressa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.77 Durban 2023
http://dx.doi.org/10.17159/0379-4350/2023/v77a20
RESEARCH ARTICLE
Molecular Dynamics Simulation of Lenalidomide Interaction with CRBN Protein: A target for immunomodulatory Drugs
Arefeh Esmaeili; Mehdi Yoosefian; Mohamad Mahani
Department of Chemistry, Graduate University of Advanced Technology, Kerman, Iran
ABSTRACT
Lenalidomide, an immune-modulating drug, has shown promising therapeutic efficacy in treating haematological malignancies. Lenalidomide's primary target is Cereblon (CRBN), a widely expressed protein, which plays a crucial role and found in the CUL4-RBX1-DDB1-containing E3 ubiquitin ligase complex known as CRL4. The interaction between CRBN and DDB1 results the formation of a CUL4-based E3 ubiquitin ligase complex. This complex is responsible for the ubiquitination and proteasomal degradation of proteins identified by CRBN, thereby maintaining cellular homeostasis, survival, division, proliferation, and growth by eliminating non-essential or damaged proteins. Remarkably, lenalidomide exhibits selective prevention of substrate binding to CRBN through an anti-selective mechanism and directly binds to CRBN. This mechanism has been extensively linked with treating patients with multiple myeloma-related cancers. In this study, we employ molecular dynamics simulations to investigate the interaction of lenalidomide with the CRBN protein, the primary target of immunomodulatory drugs. Our results demonstrate the effective exchange of lenalidomide with its primary target.
Keywords: Lenalidomide, Cereblon, Ubiquitin, Substrate, Molecular Dynamics Simulations
INTRODUCTION
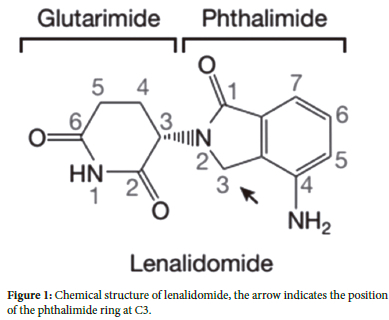
Immunomodulatory drugs (IMiDs) are a class of immune-regulating medications that contain an imide group.1,2 The IMiD class includes thalidomide and its analogs (lenalidomide, pomalidomide).3 Lenalidomide, marketed as REVLIMID,4 gained FDA approval in September 2011 as a novel thalidomide analogue.5-9 Lenalidomide is a phthalimide compound with an amino substitution at position four and a dioxopiperidin-3-yl group (a chemical functional group) at position 2 of the oxo isoindoline-1 core, which inhibits TNF-α secretion. Lenalidomide serves as an anti-angiogenic, anti-neoplastic, and immunomodulatory agent. Experts classify lenalidomide as an aromatic amine, falling under the isoindoles subclass of phthalimides and the piperidones subclass (Figure 1).
Lenalidomide, a drug with multiple mechanisms of action, including enhancing efficacy in tumour and immune cells, has been studied extensively.10,11 The immunomodulatory effects of lenalidomide include altering cytokine production, regulating the simultaneous activation of T-cells, and enhancing cellular toxicity mediated by NK cells. Lenalidomide modifies the substrate specificity of the CRL4CRBN complex to utilize ligase substrates,12 including Ikaros (IKZF1) and Aiolos (IKZF3),13,14 leading to their ubiquitination and subsequent degradation. The transcription factors Ikaros and Aiolos have been identified as substrates of the CRL4CRBN-Lenalidomide drug complex, explaining many of the therapeutic effects of IMiD compounds on immune and tumour cells.15
IKZF1 and IKZF3 are transcription factors in B cells that are essential for B cell differentiation and malignant cell survival. IKZF3, also known as Interferon 4 (IRF4) regulatory factor,16 is a transcription factor that controls the expression of specific myeloma genes. The expression of the IRF4 regulatory factor has shown a reduction in multiple myeloma cell lines and bone marrow samples from myeloma patients treated with lenalidomide. Lenalidomide can partly explain its immunomodulatory effects through the degradation of IKZF3, ultimately exhibiting pleiotropic activities.
After over a decade of investigation into the mechanisms of action of lenalidomide, preclinical studies have identified cereblon (CRBN) in the CRL4CRBN complex as a direct molecular target for lenalidomide's teratogenicity. CRBN interacts directly with lenalidomide and indirectly with DDB1, contributing to the teratogenic effects of lenalidomide. Lenalidomide initiates its teratogenic effects by binding to CRBN and inhibiting the activity of the E3 ubiquitin ligase. Recent structural studies have indicated that the interaction between lenalidomide and CRBN is predominantly mediated through the glutarimide ring. In contrast, the phthalimide ring, along with a small region of the brain surface, is involved in interactions with neo-substrates for ubiquitination within the ternary complex.17-19
Lenalidomide possesses a chiral center (C3 carbon of the glutarimide ring) (Figure 1).20 Structural and biochemical studies have demonstrated that the (S) enantiomer exhibits approximately tenfold more robust binding to the brain and more significant self-ubiquitination inhibition compared to the (R) enantiomer, which links to the teratogenic effects of the (S) isomer. The binding site of IMiD compounds is a shallow hydrophobic pocket on the surface of CRBN.
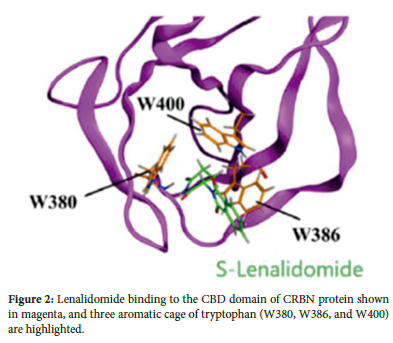
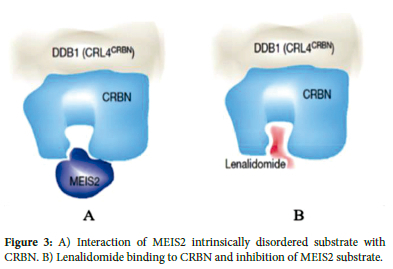
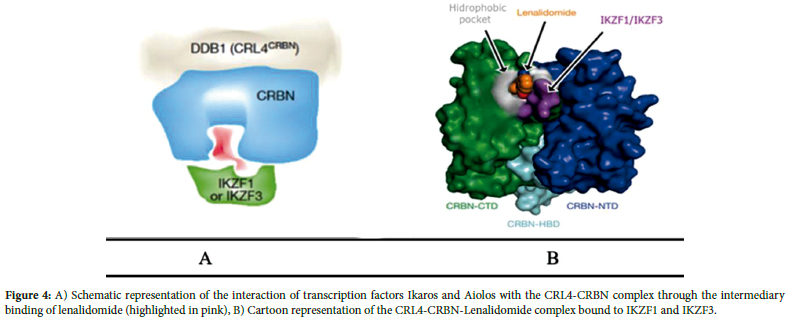
Three tryptophan residues form hydrogen bonds between CRBN and the glutarimide ring of lenalidomide (Figure 2). The glutarimide ring represents a characteristic feature of the IMiD class of molecules, and all examined CRBN-IMiD structures have shown highly similar intermolecular interactions. Specifically, lenalidomide causes the CRL4CRBN complex to target the intrinsically disordered ligand (MEIS2) for degradation (Figure 3). The binding of transcription factors Ikaros and Aiolos to CRBN requires a similar intermediate compound, as shown in Figure 4A.9 Following drug binding to CRBN, lenalidomide acts as an intermediate, leading to increased substrate binding of Ikaros and Aiolos to the E3 ubiquitin ligase complex and their subsequent degradation, with the highest substrate binding occurring through the direct interaction of lenalidomide's C4 amine and the phthalimide ring (Figure 4B).
Our main objective in this article is to investigate the binding of lenalidomide to its primary target, the CRBN protein, using molecular dynamics simulations. This approach allows us to gain insights into the dynamic behaviour of the lenalidomide-CRBN interaction, which plays a crucial role in the recruitment and subsequent degradation of lymphoid transcription factors by the CRL4CRBN ligase complex.
METHOD
Molecular dynamics simulations
In this research, we employed molecular dynamics simulations with the GROMACS program version 2022.4 to investigate the interactions between the drug and protein.21 We retrieved the crystal structure of the protein and the drug (with code 4TZ4) from the protein database. To ensure compatibility with the Charmm36 force field,22 the drug structure has been optimized. We positioned the protein at the centre of the simulation box for the molecular dynamics simulation, and introduced the drug molecule into the box. We maintained the appropriate conditions, including setting the distance between the protein-ligand complex and the box wall to 1 Á. The TIP3P We reduced the system's energy and resolved conflicts by applying an energy minimization algorithm (descent minimization steep = steepest) for 50 000 initial integration steps. We conducted temperature equilibration by performing an NVT ensemble simulation at 300K for one ns,23 employing the V-rescale thermostat. To achieve pressure equilibration, we simulated an NPT ensemble at 1 bar pressure for one ns,24 utilizing the Parrinello-Rahman barostat.25 We conducted the primary molecular dynamics simulation for 50 ns. We present the comprehensive list of all system components used in these simulations in Table 1. In this study, the primary objective was to employ molecular dynamics simulations to investigate the interactions between the protein and the drug. The analyses have been aimed to enhance our understanding of the compound's impact on lymphoid structures and tumour cells.
RESULTS
Molecular dynamics result
We conducted molecular dynamics simulations26 to analyze and investigate the inhibitory effect of lenalidomide on the Cereblon protein. We used the simulation trajectories to explore various crucial aspects, including the Root Mean Square Deviation (RMSD) of the drug at the binding site, the Root Mean Square Fluctuation (RMSF) of the protein, hydrogen bond interactions between the protein and the drug, calculation of the distance between the drug and the protein, as well as the conformational changes in active site residues and the gyration radius of Cereblon protein. We performed Solvent Accessible Surface Area (SASA) analysis to assess the exposed surface of the protein in the presence of solvent.
The results obtained from these comprehensive analyses are presented in this section of the article, offering valuable insights into the interaction mechanisms and dynamics of lenalidomide with the cereblon protein.
System optimization
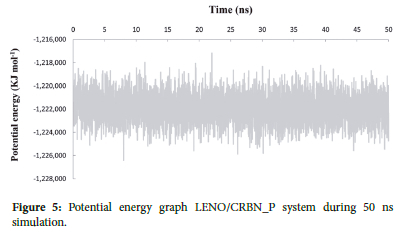
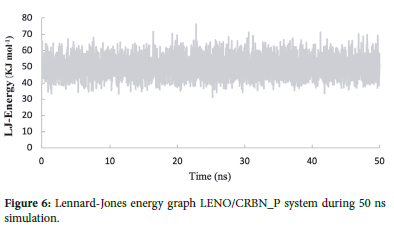
Different parameters, such as total energy and potential energy, are used as indicators to investigate the systems stability. Energy minimization aims to bring the system to its most stable state possible, eliminating spatial clashes or improper overlaps in the protein-drug complex. Once the system reaches the minimum energy state, we can initiate the simulation. Figures 5 and 6 display the profiles of potential and Lennard-Jones energy27 for the complex throughout the 50 ns simulation.
RMSD analysis
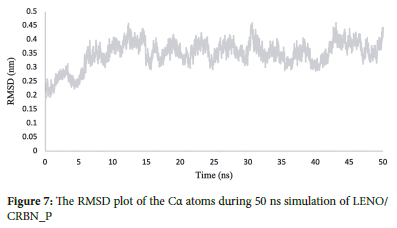
RMSD, or Root Mean Square Deviation,28 is a numerical measurement used to assess the difference between two structures: the target structure and a reference structure. In the context of molecular dynamics simulations, RMSD helps us understand how structures change over time compared to their initial state. This analysis is particularly useful for studying time-dependent motions and evaluating the degree of deviation from the starting coordinates, indicating structural stability throughout the simulation.
By calculating the RMSD of the drug at the binding site of the protein during a 50 ns simulation, we can investigate the structural stability of the drug-protein complex. The RMSD values for lenalidomide within the active site of the protein show relative constancy after the initial 15 ns with an average value of 0.35 nm. This observation suggests that the complex remains stable and in equilibrium during the simulation. For a visual representation, please refer to Figure 7, which illustrates the plot of RMSD over time.
RMSF analysis
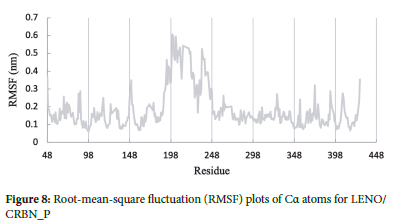
Proteins, like all molecules, undergo structural fluctuations under physiological conditions, and these fluctuations play a crucial role in the functionality of proteins across various time scales. RMSF, which stands for Root Mean Square Fluctuation,29 is a valuable tool to compute and investigate the flexibility and dynamics of protein structures upon ligand binding.
In the context of this study, Figure 8 displays the fluctuations of Ca atoms for each residue in the complexes. From the analysis, it is clear that lenalidomide does not significantly change the overall flexibility of the cereblon protein structure at the active site. The reason for this observation can be attributed to the fact that the active site residues lie between amino acids 300-400, and lenalidomide does not cause substantial changes in their dynamics. The overall structural flexibility of the cereblon protein remains relatively unaffected by the presence of lenalidomide at the active site.
H-Bond analysis
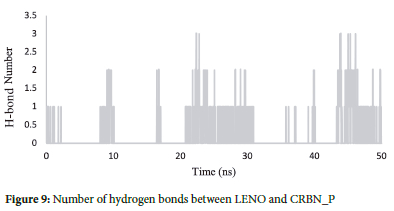
Hydrogen bonding occurs when a weakly positive hydrogen atom in one molecule is attracted to a highly electronegative atom in another.30-34 Analyzing the number of hydrogen bonds formed between the drug and the protein provides valuable insights into the drug's affinity for it. It plays a crucial role in stabilizing the drug-protein binding.
According to the results obtained in this study, there are approximately 2 to 3 hydrogen bonds between the protein and the drug. Notably, most of these hydrogen bonds are formed at the binding site, explicitly involving the Trp380(C) and His378(C) residues of the protein and the drug (Figure 9). This observation suggests that these specific residues play a significant role in forming hydrogen bonds and contribute to the stability of the drug-protein complex.
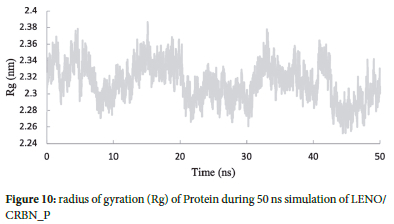
Rg analysis
Analyzing the radius of gyration (Rg) provides valuable insights into the structural characteristics of the protein-drug complex throughout the molecular dynamics (MD) simulation.35 The radius of gyration (Rg) is defined as the distribution of atoms of a protein around its axis. In this study, we calculated the Rg values for the LENO/PR complex during the 50 ns MD simulation (Figure 10). The observed average Rg value for the LENO/PR complex was approximately 2.31 nm. This value suggests that the protein-drug complex maintains a relatively compact and stable structure throughout the simulation. The consistent Rg values indicate that the complex remains in a well-defined conformation, demonstrating its structural stability for simulation.
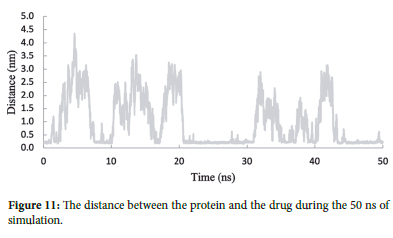
Distance analysis
The fluctuation in the distance between the protein and the drug over time is a crucial parameter to assess the stability of the protein-drug complex. When the length increases, it suggests that the protein and the drug are not interacting strongly, and the complex may be less stable. Conversely, when the distance remains relatively constant, the protein and the drug maintain a regular interaction throughout the simulation.
Analyzing the fluctuations in the protein-drug distance helps researchers understand the dynamic nature of the protein-drug complex and its binding interactions.36 The constant distance observed at certain time intervals may indicate the formation of stable binding pockets or hydrogen bonds between the protein and the drug, contributing to the overall stability of the LENO/PR complex.
Figure 11 illustrates the distance between the protein and the drug during the 50 ns simulation. According to the Figure 11, a significant peak is observed for the LENO/PR complex, indicating that the protein and the drug have moved away, resulting in fewer contacts between them. However, at different time points, the distance between LENO/CRBN_P remains relatively constant, around 0.2 nm.
Number of contact analysis
The contact analysis between lenalidomide (LENO) and CRBN protein (CRBN_P) was performed to investigate the contacts between these two molecules during the molecular dynamics simulation. Figure 12 illustrates the changes in contacts over 50 ns of simulation time. At the beginning of the simulation, when LENO and CRBN_P are initially distant from each other, the number of contacts is relatively low. As time progresses, the interactions between LENO and CRBN_P increase, leading to a rise in the number of contacts.
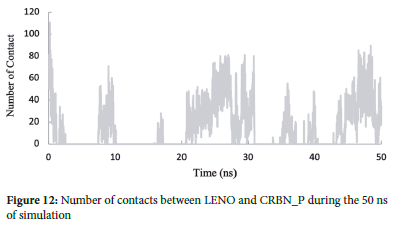
Between 20 to 30 ns, the number of contacts reaches its maximum value of 80, indicating a substantial level of interaction between LENO and CRBN_P. This period represents a significant and robust binding event between the drug and the protein, where they establish multiple contacts.
The decrease in the number of contacts after the peak could be due to conformational changes in the complex or temporary dissociation of the drug from the binding site. Despite the fluctuations in the number of contacts, the interaction between LENO and CRBN_P remains present throughout the simulation, showcasing the dynamic nature of their binding.
This contact analysis provides valuable insights into the specific residues and regions of the protein involved in the interactions with the drug, shedding light on the crucial binding events that underlie the inhibitory effect of lenalidomide on the CRBN protein.
SASA analysis
The SASA (solvent-accessible surface area) analysis offers valuable insights into the surface properties of the LENO/CRBN_PR complex and its interactions with the surrounding solvent, providing a better understanding of potential functional roles and structural changes induced by solvation effects during the molecular dynamics simulation.37 As depicted in Figure 13, the solvent-accessible surface area (SASA) for the LENO/CRBN_PR complex is approximately 200 nm2. SASA (solvent-accessible surface area) is a measure of the surface area of a molecule that is accessible to the solvent. It provides valuable information about the exposed regions of the protein and the drug in the complex.
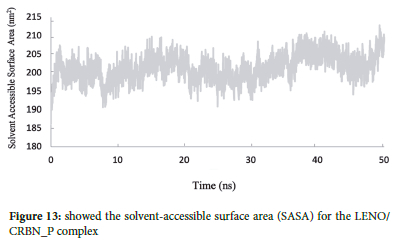
The observed solvent-accessible surface area of 200 nm2 indicates that a significant portion of the LENO/CRBN_PR complex is exposed to the surrounding solvent. This suggests that the complex has regions accessible for interactions with solvent molecules. The interaction with the solvent may influence the conformational changes and behaviour of the complex, potentially affecting its overall function and behaviour in the biological environment.
DISCUSSION
The molecular dynamics simulations of the LENO/CRBN_P complex have provided valuable insights into its structural and dynamic behaviour. The analysis of various parameters, such as RMSD, RMSF, hydrogen bond interactions, distance calculations, and solvent-accessible surface area (SASA), has shed light on the interactions between lenalidomide (LENO) and the CRBN protein (CRBN_P).
The RMSD analysis revealed that the LENO/CRBN_P complex reached a stable state after approximately 20 ns of simulation, indicating that the complex achieved equilibrium and maintained a relatively constant overall structure during the simulation.
The RMSF analysis showed that lenalidomide in the active site of the CRBN protein did not significantly affect the overall flexibility of the protein. Results show that the binding of lenalidomide did not induce significant structural changes in the CRBN protein, and the complex remained stable throughout the simulation.
The analysis of hydrogen bond interactions between LENO and CRBN_P demonstrated that the complex formed between 2 to 3 hydrogen bonds. These hydrogen bonds are crucial for stabilizing the drug-protein interaction, and their presence indicates the favourable affinity of lenalidomide for the CRBN protein.
The distance analysis showed that lenalidomide and the CRBN protein exhibited close interactions during the early stages of the simulation. The number of contacts increased to a maximum value of 80 within 20 to 30 ns, indicating significant interactions between the two molecules. However, after this time, the number of contacts gradually decreased, suggesting a reduction in clashes or steric hindrances between the drug and the protein.
Moreover, the solvent-accessible surface area (SASA) analysis provided an estimate of the surface area exposed to the solvent by the LENO/CRBN_P complex. The calculated SASA value of approximately 200 nm2 indicates a substantial portion of the complex is accessible to the solvent. This exposure to solvent molecules can influence the dynamics and stability of the complex and potentially modulate its functional properties.
CONCLUSION
In this study, we investigated the interactions between the drug lenalidomide and the CRBN protein using molecular dynamics simulations. We simulated the drug-protein interaction in an aqueous solvent and NPT ensemble with the CHARMM36 force field for 50 ns.
Based on the results, we can infer that lenalidomide binds to its primary target, the CRBN protein. It exhibits desirable interactions and can potentially form the CRL4CRBN-Lenalidomide complex to recruit transcription factors Ikaros and Aiolos. These findings contribute to a better understanding of the drug-protein interaction and its potential implications in targeted therapies for various diseases. However, further experimental studies are warranted to validate and expand the simulation results, thus enhancing our knowledge of this drug-protein complex and its biological implications.
ACKNOWLEDGEMENTS
With gratitude and appreciation to the Graduate University of Advanced Technology in Kerman, Iran, for their valuable support and facilitation provided during the research for this article.
ORCID IDS
Arefeh Esmaeili: https://orcid.org/0009-0003-5472-7301
Mehdi Yoosefian: https://orcid.org/0000-0003-0096-5083
Mohammad Mahani: https://orcid.org/0000-0001-5258-8561
REFERENCES
1. Raza S, A Safyan R, Lentzsch S. Immunomodulatory drugs (imids) in multiple myeloma. Curr Cancer Drug Targets. 2017;17(9):846-857. https://doi.org/10.2174/1568009617666170214104426 [ Links ]
2. Knight R. Imids: A novel class of immunomodulators. Semin Oncol. 2005: Elsevier. https://doi.org/10.1053/j.seminoncol.2005.06.018 [ Links ]
3. Franks ME, Macpherson GR, Figg WD. Thalidomide. Lancet. 2004; 363(9423):1802-1811. https://doi.org/10.1016/S0140-6736(04)16308-3 [ Links ]
4. Zhu D, Corral LG, Fleming YW, Stein B. Immunomodulatory drugs revlimid*(lenalidomide) and cc-4047 induce apoptosis of both hematological and solid tumor cells through nk cell activation. Cancer Immunol Immunother. 2008;57:1849-1859. https://doi.org/10.1007/s00262-008-0512-7 [ Links ]
5. Aragon-Ching JB, Li H, Gardner ER, Figg WD. Thalidomide analogues as anticancer drugs. Recent Pat Anticancer Drug Discov. 2007;2(2):167-174. https://doi.org/10.2174/157489207780832478 [ Links ]
6. Raedler LA. Revlimid (lenalidomide) now fda approved as first-line therapy for patients with multiple myeloma. Am Health Drug Benefits. 2016;9(Spec Feature):140. [ Links ]
7. Dredge K, Marriott J, Macdonald C, Man H, Chen R, Muller G, Stirling D, Dalgleish A. Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br J Cancer. 2002;87(10):1166-1172. https://doi.org/10.1038/sj.bjc.6600607 [ Links ]
8. Beedie SL, Peer CJ, Pisle S, Gardner ER, Mahony C, Barnett S, Ambrozak A, Gütschow M, Chau CH, Vargesson N. Anticancer properties of a novel class of tetrafluorinated thalidomide analogues. Mol Cancer Ther. 2015;14(10):2228-2237. https://doi.org/10.1158/1535-7163.MCT-15-0320 [ Links ]
9. Teo SK, Stirling DI, Zeldis JB. Thalidomide as a novel therapeutic agent: New uses for an old product. Drug Discov Today. 2005;10(2):107-114. https://doi.org/10.1016/S1359-6446(04)03307-0 [ Links ]
10. Zeldis JB, Knight R, Hussein M, Chopra R, Muller G. A review of the history, properties, and use of the immunomodulatory compound lenalidomide. Ann N Y Acad Sci. 2011;1222(1):76-82. https://doi.org/10.1111/j.1749-6632.2011.05974.x [ Links ]
11. Richardson P, Mitsiades C, Laubach J, Schlossman R, Ghobrial I, Hideshima T, Munshi N, Anderson K. Lenalidomide in multiple myeloma: An evidence-based review of its role in therapy. Core Evid. 2009;4:215. https://doi.org/10.2147/ce.s6002 [ Links ]
12. Barankiewicz J, Salomon-Perzynski A, Misiewicz-Krzeminska I, Lech-Maranda E. Crl4crbn e3 ligase complex as a therapeutic target in multiple myeloma. Cancers. 2022;14(18):4492. https://doi.org/10.3390/cancers14184492 [ Links ]
13. Olsson L, Johansson B. Ikaros and leukaemia. Br J Haematol. 2015;169(4):479-491. https://doi.org/10.1111/bjh.13342 [ Links ]
14. Bjorklund C, Lu L, Kang J, Hagner P, Havens C, Amatangelo M, Wang M, Ren Y, Couto S, Breider M. Rate of crl4crbn substrate ikaros and aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-myc and irf4. Blood Cancer J. 2015;5(10):e354-e354. https://doi.org/10.1038/bcj.2015.66 [ Links ]
15. Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM. Structure of the ddb1-crbn e3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49-53. https://doi.org/10.1038/nature13527 [ Links ]
16. Maffei R, Fiorcari S, Benatti S, Atene CG, Martinelli S, Zucchini P, Potenza L, Luppi M, Marasca R. Irf4 modulates the response to bcr activation in chronic lymphocytic leukemia regulating ikaros and syk. Leukemia. 2021;35(5):1330-1343. https://doi.org/10.1038/s41375-021-01178-5 [ Links ]
17. Ito T, Ando H, Handa H. Teratogenic effects of thalidomide: Molecular mechanisms. CMLS. 2011;68:1569-1579. https://doi.org/10.1007/s00018-010-0619-9 [ Links ]
18. Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu C-C, Miller K, Fang W, Wang N-Y, Nguyen D, Houston J. A novel cereblon modulator recruits gspt1 to the crl4crbn ubiquitin ligase. Nature. 2016;535(7611):252-257. https://doi.org/10.1038/nature18611 [ Links ]
19. Petzold G, Fischer ES, Thomâ NH. Structural basis of lenalidomide-induced ck1a degradation by the crl4crbn ubiquitin ligase. Nature. 2016;532(7597):127-130. https://doi.org/10.1038/nature16979 [ Links ]
20. Eriksson T, Bjöurkman S, Roth B, Fyge Á, Höuglund P. Stereospecific determination, chiral inversion in vitro and pharmacokinetics in humans of the enantiomers of thalidomide. Chirality. 1995;7(1):44-52. https://doi.org/10.1002/chir.530070109 [ Links ]
21. Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19-25. https://doi.org/10.1016/j.softx.2015.06.001 [ Links ]
22. Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, De Groot BL, Grubmüller H, MacKerell Jr AD. Charmm36m: An improved force field for folded and intrinsically disordered proteins. Nat Methods. 2017;14(1):71-73. https://doi.org/10.1038/nmeth.4067 [ Links ]
23. Andersen HC. Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys. 1980;72(4):2384-2393. https://doi.org/10.1063/L439486 [ Links ]
24. Heyes D. Molecular dynamics at constant pressure and temperature. Chem Phys. 1983;82(3):285-301. https://doi.org/10.1016/0301-0104(83)85235-5 [ Links ]
25. Saito H, Nagao H, Nishikawa K, Kinugawa K. Molecular collective dynamics in solid para-hydrogen and ortho-deuterium: The parrinello-rahman-type path integral centroid molecular dynamics approach. J Chem Phys. 2003;119(2):953-963. https://doi.org/10.1063/L1578474 [ Links ]
26. Hansson T, Oostenbrink C, van Gunsteren W. Molecular dynamics simulations. Curr Opin Struct Biol. 2002;12(2):190-196. https://doi.org/10.1016/S0959-440X(02)00308-1 [ Links ]
27. Van der Hoef MA. Free energy of the lennard-jones solid. J Chem Phys. 2000;113(18):8142-8148. https://doi.org/10.1063/L1314342 [ Links ]
28. Sargsyan K, Grauffel C, Lim C. How molecular size impacts rmsd applications in molecular dynamics simulations. J. Chem. Theory Comput. 2017;13(4):1518-1524. https://doi.org/10.1021/acs.jctc.7b00028 [ Links ]
29. Martinez L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PloS ONE. 2015;10(3):e0119264. https://doi.org/10.1371/journal.pone.0119264 [ Links ]
30. Buckingham A, Del Bene J, McDowell S. The hydrogen bond. Chem Phys Lett. 2008;463(1-3):1-10. https://doi.org/10.1016/j.cplett.2008.06.060 [ Links ]
31. Raissi H, Nadim ES, Yoosefian M, Farzad F, Ghiamati E, Nowroozi AR, Fazli M, Amoozadeh A. The effects of substitutions on structure, electron density, resonance and intramolecular hydrogen bonding strength in 3-mercapto-propenethial. Theochem. 2010;960(1-3):1-9. https://doi.org/10.1016/j.theochem.2010.08.012 [ Links ]
32. Raissi H, Yoosefian M, Mollania F, Farzad F, Nowroozi AR. Ab initio and dft computational studies on molecular conformations and strength of the intramolecular hydrogen bond in different conformers of 3-amino-2-iminomethyl acryl aldehyde. Comput Theor Chem. 2011;966(1-3):299-305. https://doi.org/10.1016/j.comptc.2011.03.026 [ Links ]
33. Raissi H, Yoosefian M, Mollania F, Khoshkhou S. Electronic structures, intramolecular interactions, and aromaticity of substituted 1-(2-iminoethylidene) silan amine: A density functional study. Struct Chem. 2013;24:123-137. https://doi.org/10.1007/s11224-012-0038-7 [ Links ]
34. Raissi H, Yoosefian M, Mollania F. Hydrogen bond studies in substituted imino-acetaldehyde oxime. Comput Theor Chem. 2012;996:68-75. https://doi.org/10.1016/j.comptc.2012.07.017 [ Links ]
35. Lobanov MY, Bogatyreva N, Galzitskaya O. Radius of gyration as an indicator of protein structure compactness. Mol Biol. 2008;42:623-628. https://doi.org/10.1134/S0026893308040195 [ Links ]
36. Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O. Mdanalysis: A toolkit for the analysis of molecular dynamics simulations. J Comput Chem. 2011;32(10):2319-2327. http://doi.org/10.1002/jcc.21787 [ Links ]
37. Tanner DE, Phillips JC, Schulten K. Gpu/cpu algorithm for generalized born/solvent-accessible surface area implicit solvent calculations. J. Chem. Theory Comput. 2012;8(7):2521-2530. http://doi.org/10.1021/ct3003089 [ Links ]
Received 29 July 2023
Revised 6 September 2023
Accepted 7 September 2023
* To whom correspondence should be addressed. Email: myoosefian7@gmail.com


{kind=link}