










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Chemistry
versão On-line ISSN 1996-840X
versão impressa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.77 Durban 2023
http://dx.doi.org/10.17159/0379-4350/2023/v77a13
RESEARCH ARTICLE
Computational study on the antioxidant activity of five plant food benzoic acid derivatives: dearomatization and stability of H abstraction radicals
Bienfait K. IsamuraI, II, *; Issofa PatouossaIII; Isaac E. KabaIV; Aristote MatondoII; Pius T. MpianaII
IDepartment of Chemistry, Rhodes University, Makhanda, South Africa
IIDepartment of Chemistry, Faculty of Sciences, University of Kinshasa, Kinshasa XI, Democratic Republic of the Congo
IIIPhysical and Theoretical Chemistry Unit, Laboratory of Applied Physical and Analytical Chemistry, Faculty of Sciences, University of Yaoundé I, Yaoundé, Cameroon
IVDepartment of Medicinal and Biological Chemistry, College of Pharmacy and Pharmaceutical Sciences, The University of Toledo, Toledo, Ohio, United States of America
ABSTRACT
In this study, we report a computational investigation of the antioxidant activity of five plant food benzoic acid derivatives, namely gallic acid (GA), para-hydroxybenzoic acid (PHBA), protocatechuic acid (PCA), syringic acid (SA), and vanillic acid (VA). Based on a series of computed thermodynamics parameters, we have developed a comparative debate regarding their free radical scavenging activity in the gas phase and polar solutions (considering water and methanol solvents). This discussion expands to the elucidation of both the most preferred mechanism and the order of antioxidant activity in each environment. Paradoxically, calculations using the harmonic oscillator model of aromaticity (HOMA) suggest that H abstraction radicals gain stability as the central benzene ring loses structural aromaticity. Finally, spin densities and Fukui function f0 appear to be good indicators of the local reactivity of the five antioxidants toward free radicals.
Keywords: antioxidant activity, dearomatization, frontier molecular orbital, Fukui function f0, DFT
INTRODUCTION
Antioxidant activity is one of the most important properties of food plants.1 An antioxidant is any substance occurring at lower concentrations than that of an oxidizable substrate, and capable of significantly delaying or even preventing the oxidation of that substrate.2 The human body, in fact, produces powerful antioxidants such as alpha-lipoic acid and glutathione.3 However, these natural "soldiers" may not be at the level of the expectations. Therefore, it is often recommended to regularly supply the body with external sources of antioxidants.4 Antioxidants are naturally found in plants,5-8 animals,9,10 and microorganisms 11,12 or they may be synthesized by chemical means13,14. With the perpetual need to identify more effective antioxidant candidates to integrate into the preventive or therapeutic pipeline of several pathologies, the isolation and synthesis of new antioxidants is a hot research domain attracting the interest of a myriad of scientists.15-18
Over the past decades, numerous studies have documented the antioxidant activity of benzoic acid derivatives (BADs). To cite only a few, Velika et al. 19 experimentally studied the antioxidant activity of fourteen benzoic acid derivatives against superoxide radicals using the Beauchamp-Fridovick method. Their study revealed that monohydroxybenzoic derivatives had the best antioxidant power. The same authors also noticed that molecules bearing the -OH group in ortho and para positions to the carboxylic group showed the highest activity. On the same track, Ali-Haidari et al. 20 isolated a series of benzoic acid derivatives from Cassia itálica, a plant growing in South Arabia, and showed them to display considerable antioxidant activities in DPPH assays, reaching up to 95.8% as compared to butylated hydroxyanisole (~94%). Furthermore, keeping in mind the need to identify potential natural substances that can reduce the severity of alcoholic liver disease (ALD), a disease known to be related to oxidative stress, several investigations have been conducted and many more are still popping up. In this vein, Saravanan and coworkers showed that 2-Hydroxy-4-methoxy benzoic acid (HMBA), the active principle of Hemidesmus indicus, significantly inhibits the development of liver injury in ethanol administration.21
Besides experimental works, the literature also documents a panoply of theoretical studies that have contributed to unveiling the magic behind the antioxidant properties and site selectivity of many BADs. For instance, the homolytic and heterolytic splitting-off of the phenolic -OH groups of gallic acid and its carboxylic anion in gas-phase, benzene, and water solutions were elucidated by Skorna et al. 22 using the B3LYP functional combined with the 6-311++G (d,p) basis set. These authors found that the deprotonation of the carboxylic groups increases the antioxidant activity in all environments. Another outstanding DFT study was conducted in the gas phase to examine the effect of intramolecular hydrogen bonding and the presence of bulky fragments near the -OH group on the antioxidant activity of a series of vitamins and phenolic acids, including syringic and vanillic acids. The calculations performed suggested that intramolecular H-bonds involving ortho-hydroxy groups resulted in higher antioxidant power by lowering bond dissociation enthalpies and ionization potentials.23 In 2012, Kalita et al. 24 excellently studied the antioxidant activity of gallic acid derivatives. Their work brought unquestionable theoretical evidence that reactivity descriptors derived from conceptual density functional theory can be used to gain insights into the local reactivity of these antioxidants.
Furthermore, the literature indicates that phenolic antioxidants ArO-H protect against oxidative stress according to three possible mechanisms (Figure 1). In the first model, known as Hydrogen Atom Transfer (HAT), ArO-H donates an H atom to a reactive free radical R-, which is then converted into a neutral species RH alongside a less reactive ArO- radical.23 The second protocol is the Stepwise Electron Transfer Proton Transfer (SET-PT) pathway, where ArO-H consecutively withdraws one electron and a proton onto the free radical, leading to the same products as in the HAT route.25 The third model [Sequential Proton Loss Electron Transfer (SPLET)] involves the heterolytic dissociation of an O-H bond, followed by the subsequent shift of a single electron from ArO- to the free radical.26 Although the net result of these three mechanisms is the same, i.e. the formation of less harmful radicals, it has been shown that under certain conditions one of them always prevails.7 Following the HAT mechanism, one can assess the antioxidant activity of a given molecule ArO-H by computing the bond dissociation enthalpies (BDE) of all the O-H bonds in the molecule. This allows for the measurement of the amount of energy required to break a bond in a homolytic fashion so that small BDEs can be related to higher antioxidant activity.5 In the context of the SET-PT mechanism, the debate is grounded on two indicators, namely the ionization potential (IP) and proton dissociation enthalpy (PDE). Low IPs and PDEs are generally associated with great antioxidant properties.27 Finally, regarding the SPLET protocol, the proton affinity (PA) and the electron transfer enthalpy (ETE) are the most relied on thermodynamics parameters to discuss the antioxidant power of phenolic antioxidants. A rule of thumb is that the lower the PAs and ETEs are, the higher the antioxidant activity.28
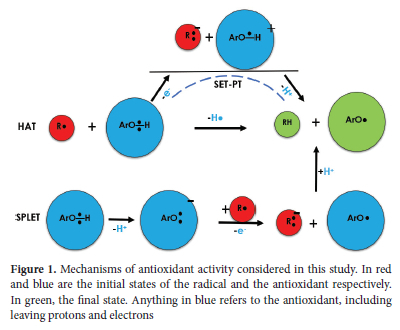
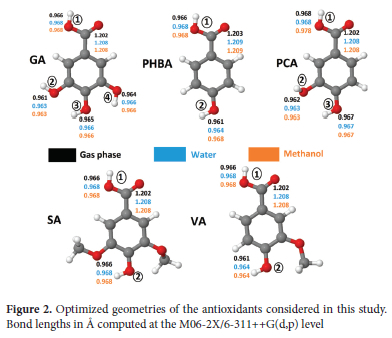
In the present study, we report an in-silico investigation of the antioxidant activity of five BADs using the M06-2X and B3LYP functionals combined with the 6-31++G(d,p) and 6-311++G(d,p) basis sets. As shown in Figure 2, these BADs comprise gallic acid (GA), para-hydroxybenzoic acid (PHBA), protocatechuic acid (PCA), syringic acid (SA), and vanillic acid (VA). All of these compounds exist naturally in plant food as components of complex structures like lignins and hydrolyzable tannins or attached to cell walls and proteins.29,30 Although their antioxidant activity had been the subject of numerous experimental works,31-33 it was time to provide further theoretical support to the empirical data accumulated so far. This study aims to elucidate the order of antioxidant activity of the five BADs following each of the HAT, SET-PT, and SPLET mechanisms and comprehend the origin of their different activities. Let us note that gallic acid is widely praised for its outstanding antioxidant activity. This molecule, nicknamed the molecular rival of cancer by Verma et al.34, is abundantly found in tomatoes, grapes, berries, and several other fruits.35,36 It is also one of the constituents of several hardwood plant species.38,39 Apart from its antioxidant activity, gallic acid and its derivatives also exhibit anti-inflammatory,40 anti-mutagenic,41 and anticancer activities.42 To account for the strong dependence of antioxidant properties on the medium where the system is found, we consider in this study three environments, i.e., gas phase, water, and methanol solutions. We begin the investigation by giving a small talk on the optimized geometries of the five antioxidants as suggested by the M06-2X/6-311++G(d,p) level. Then, their antioxidant properties are compared based on thermodynamic parameters derived from the HAT, SET-PT, and SPLET mechanisms. The data collected is then examined to postulate the most preferred pathway in the gas phase and polar solutions, as well as the most plausible order of antioxidant activity in each medium. We have also attempted to understand the effect of the H abstraction on the aromaticity of the benzene ring in terms of changes in bond lengths, using the Harmonic Oscillator Model of Aromaticity (HOMA). Finally, the reactivity of these molecules towards free radicals is debated using frontier molecular orbitals and the Fukui function f0. All these tools are known to be effective in elucidating the reactivity of molecular systems.
COMPUTATIONAL DETAILS
All the structures, i.e. neutral molecules, radicals, cations, and anions, were fully optimized using the meta hybrid M06-2X functional in conjunction with the 6-311++G(d,p) basis set as implemented in Gaussian 09 software.43 Optimized geometries are provided in Table S1 in the supporting information. The choice of the M06-2X/6-311++G(d,p) level of theory was motivated by the fact that it has been shown to provide reliable results about the antioxidant activities of many families of compounds.44 Vibrational frequency calculations confirmed the predicted stationary point as being real minima on the potential energy surface. Bond dissociation enthalpies (BDE), ionization potentials (IPs), Proton Affinities (PA), proton dissociation enthalpies (PDEs), and electron transfer enthalpies (ETEs) were calculated using equations 1-5, where Ar-OH is the neutral form of the antioxidant, ArO* its H abstracted radical, ArO+* and ArO - its radical cation and anion. Formation enthalpies for the proton and electron in the gas phase were fixed at 1.483 and 0.752 kcal mol-1 as obtained by Bartmess through the numerical solution of Fermi-Dirac statistical mechanics equations.45 Their solvation enthalpies in water and methanol at the M06-2X/6-311++G(d,p) level were retrieved from a benchmarking study by Rimarcík and coworkers.46 Note that solvent contributions to the total enthalpies were accounted for using the integral equation formalism IEF-PCM method 47,48 with default settings at 298.15 K and 1 atmosphere. This solvation model is not only computationally affordable but also known to provide acceptably accurate results.49
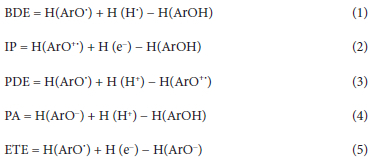
PHBA, PCA, SA, and VA. HOMA is a structural aromaticity index defined by Kruszewski and Krygowski 50 based on the idea that, in aromatic rings, bonds tend to equalize their lengths. Despite some criticism around this descriptor, many researchers have relied on it to investigate the aromaticity of a variety of systems such as polycyclic aromatic hydrocarbons and fullerenes.50 In the present case, equation 6 is employed to estimate the HOMA, with n = 6 the number of bonds forming the ring, an empirical constant equal to 257.7 for C-C bonds, and Ropt an optimal bond length valued at 1.388A and chosen so that HOMA varies between 0 and 1.
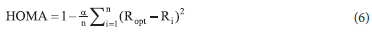
To understand the local and global reactivity of the five benzoic acid derivatives, their frontier molecular orbitals and the free radical form of the fukui function f0 were computed at the B3LYP/6-31+G(d,p) level. Be reminded that f0 is a conceptual DFT descriptor estimated as the arithmetic average of the electrophilic f-(r) and nucleophilic f+(r) fukui functions proposed by Parr et al. 51 to quantify the response of a molecular system to the fluctuation of its electron density with the respect to the number of electrons. The radical fukui function has been employed in many previous studies to unveil the local reactivity of molecular systems towards radical attacks. Typical examples include the prediction of the preferred free radical binding sites of some lignin precursors52 and the innate C-H radical functionalization sites on multi-nitrogen-containing fused arenes.53
All the structures and molecular orbitals were visualized using Jmol54 and VMD55. Isosurfaces of radical fukui functions were generated using the MultiWFN code56, while atomic condensed values were calculated using NBO charges.57
RESULTS AND DISCUSSION
Geometry and local aromaticity
Figure 2 presents optimized geometries of neutral molecules in gas phase, water and methanol solutions, as well as the convention used herein to differentiate the -OH groups of each molecule. It also indicates O-H and C=O bond lengths predicted in the three environments. Owing to the distinct electronic surroundings of OH groups in these molecules, they should exhibit different reactivity towards free radicals. On this matter, most studies agree on the fact that the greatest reactivity of benzoic acid derivatives is held in the -OH group in the para position of the carboxylic group.58 This question is re-examined later. Looking first at the geometry of these molecules, Figure 2 shows that carboxylic O-H bonds 1 are the longest, measuring between 0.961 and 0.968A in the gas phase, and between 0.968 and 0.978A in polar solution. The longest O-H bonds 1 are found in PCA regardless of the environment. Also, notice that meia-positioned O-H bonds are slightly shorter than para ones. Furthermore, the length of C=O bonds changes from 1.202-1.203A to 1.208-1.209A when going from a gas phase to polar solutions (H2O and MeOH). Altogether, these structural features suggest that both the O-H and C=O are respectively subjected to elongations of up to 0.010A and 0.006A when these molecules are dissolved in water or methanol. This observation makes sense because, even in the more realistic situation of explicit solvation where water or methanol molecules would be filled in a cage containing each of these antioxidants, one should expect intermolecular O-H...O and C=O...H hydrogen bonds to be formed between the solute and solvating molecules, resulting in similar bond stretching.59 However, the computational cost associated with such simulations constitutes a limiting factor.
HAT mechanism
Table 1 collects calculated BDEs of the five antioxidants in gas, H2O, and MeOH phases. These BDEs range between 78.2 to 111.9 kcal mol-1, whose magnitude accords with previous theoretical studies on similar systems.60,61 In the gas phase, the lowest BDE is associated with the OH group 3 of PCA, while the highest corresponds to the carboxylic OH group 1 of VA. Based on the lowest BDEs values, one could advance the following order of antioxidant activity in the gas phase: PHBA < SA < VA < GA < PCA. However, one must be cautious here because the tiny gaps between BDE values fall within the limits of accuracy of our level of computation. In addition, notice that the OH group in para position with respect to the carboxylic group exhibits the lowest BDEs in all the compounds and should, therefore, be the most active in the gas phase through the HAT pathway. Turning to the results in solutions, Table 1 reveals that the hydrogen abstraction ability of these compounds is only slightly affected by the two polar solvents, with deviations representing less than 10% of the dissociation enthalpy in the gas phase. This observation corroborates with previous findinds by Skorna and co-workers, who found that the BDEs of gallic acid O-H groups vary between 1.0 and 3.7% when going from gas phase to aqueous solution.22 From BDE values, GA could be the most active in H2O and MeOH solutions, with the lowest BDE of 78.2 and 80.4 kcal mol-1 respectively in water and methanol attributed to the OH group 3. These values corroborate with experimental data reported by Denisova et al.62, and Alberti et al.63, who found BDEs of 83.03 and 81.00 kcal mol-1 in methyl linoleate and acetonitrile/chlorobenzene respectively. In polar solutions, the trend of antioxidant activity seems slightly different as GA gets more active than PCA [PHBA < SA < VA < PCA < GA]. In all three environments, PHBA is predicted as the least active if the HAT mechanism is concerned.
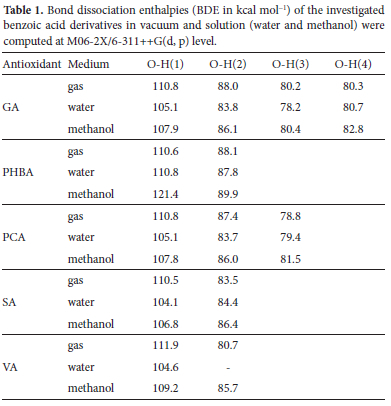
Although BDEs values clearly point out the most active site and compounds in gas (and polar phases), they do not tell why this should be so. Fortunately, common sense suggests that, after homolytic breakage of an O-H bond, the single electron left on the O atom delocalizes over the molecule and one can assume that the more it spreads away from its initial position, the higher the stability of the radical and the lower should be the BDE. Therefore, as a first approach, it is possible to rely on the distribution of spin densities to interpret the trends in BDE values.64 In our present case, spin density distributions of the radicals were calculated at the M06-2X/6-311++G (d, p) level and are here listed in Table 2. Inspection of this table suggests that for the same compound, the OH group in the para position (i.e., position 3 for GA and PCA, and position 2 for PHBA, SA and VA) has the lowest spin density estimated at 0.66, 0.67. 0.68. 0.71 and 0.61 respectively for GA, PHBA, PCA, SA, and VA, and should lead to the most stable radical. This observation is consistent with the BDEs listed in Table 1. Similarly, the highest spin density of 0.96-0.98 predicted for the carboxylic OH group corroborates well with the high BDEs reported in Table 1.
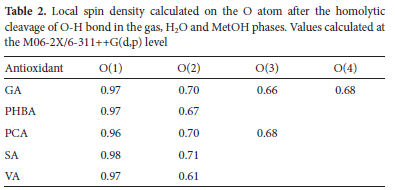
Some studies have shown that the stability order of H abstraction radicals can be regarded as the consequence of the number of resonance forms pointing to the same particular hybrid, assuming the conjugation contribution to be essentially the same for all the considered systems.65,66 However, we can admit that counting resonance structures is not a reliable nor systematic approach. Doubtlessly, once the radical is formed, its unpaired electron delocalizes over the benzene ring. This redistribution of the internal electron density produces considerable structural changes, which affect the original aromaticity of the ring. To test this assumption (in the gas phase) and find the link between the stability of H abstraction radicals and the change in aromaticity, we computed HOMA values related to the benzene ring in parent molecules and all the radicals originating from the removal of one H atom (Figure 2). Figure S1 in the supplementary information shows HOMA values of the benzene ring in all the parent molecules and the resulting radicals. The same data is summarized in Table 3. Keep in mind that lower HOMA values denote reduced geometrical aromaticity of the ring. Table 3 indicates that HOMAs are comprised between 0.980 and 0.993 for parent molecules, which denotes strong aromaticity. When an H atom is removed from one of the O-H groups, these values generally decrease and denote the loss of aromaticity by the ring. More importantly, the higher the dearomatization, the higher the stability of the radical. Therefore, induced dearomatization is the most pronounced when the H atom is expelled from the O-H group in the para position. For instance, in the case of GA, radical 3 has the lowest HOMA of 0.499, followed by radical 4 (O.542), radical 2 (0.623), and radical 1 (0.993), while the order of stability of these radicals is as follows: radical 3 > radical 4 > radical 2 > radical 1. This paradoxical observation may be understood in terms of compensation: radicals being naturally unstable, the loss of aromatic stability tries to compensate over its intrinsic instability. All these observations are in par with the findings reported in a recent study (2021) by Lin et al. 67 who used various aromaticity indices to systematically investigate the relationship between aromaticity and the thermodynamic stability of a-methyl heterocyclics. In sharp contrast with the general knowledge that aromaticity is the carrier of thermodynamic stability, their calculations showed that the stronger the antiaromaticity of the original form heterocyclics, the higher the thermodynamic stability of the corresponding radicals.
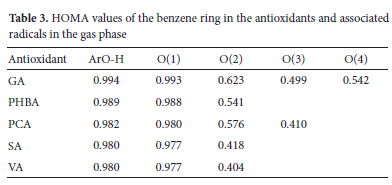
However, it must be noted that the previous rule (i.e., the higher the dearomatization, the more stable the radical) as well as the analysis of spin densities are only valid when it comes to predicting the most active site within the same molecule and should not be used to compare radicals emerging from different molecules. To illustrate this, let us consider VA and PCA. Figure 2 shows that the HOMA of radical 2 of VA is 0.404 while that for radical 3 of PCA is 0.410. In contrast, the homolytic rupture of the H atom from the O3-H group of PCA requires less energy than the one needed to split-off O2-H in VA. Similarly, spin densities at O3 (PCA) and O2(VA) are respectively 0.67 and 0.61, while corresponding BDEs are estimated to be 78.8 and 80.7 kcal mol-1 respectively in the gas phase. Similar conclusions can be made in solutions. In section 3.6, the same question is addressed from a completely different angle that consists in predicting the O atom that is the most likely to interact with an approaching free radical.
SET-PT mechanism
Table 4 reports the calculated IPs and PDEs of the five antioxidants in the gas phase and H2O and MeOH solutions at the M06-2X/6-311++G(d,p) level. The data in this table reveal that the IP values range between 185.5 and 195.5 kcal mol-1 in the gas phase, and between 121.8 and 132.9 kcal mol-1, and 127.1 and 138.3 kcal mol-1 respectively in water and MeOH. These values are in the same range as those obtained in previous studies.65,68 As far as the SET-PT model is concerned, the lowest IP values suggest the following orders of antioxidant activity: PHBA < PCA~GA< VA < SA, PHBA < GA~PCA < VA < SA, and PHBA < VA < GA~PCA < SA respectively in the gas phase, H2O and MeOH. These patterns are far different from the ones obtained with BDEs. In order words, if the reaction had to follow this pathway, then the order of antioxidant activity should be different from the one inferred from the analysis of BDEs. This apparent discrepancy between IPs and BDEs is not surprising. It may be related to the fact that BDEs are local quantities depending on the relative positions of substituents, whereas IPs are global parameters affected by the structure of the entire molecule.69 Furthermore, it is worth noting that IPs decrease drastically from the gas phase to pta M06-2X/6-311++G(d,p) olar media. For instance, the IP of SA plummets from 185.5 to 124.8 kcal mol-1 on going from the gas phase to the aqueous solution. This shows that polar solvents considerably facilitate the electron transfer to the free radicals and thus enhance the antioxidant activity. This result is in agreement with many documented studies on antioxidant properties.70 Notice also that IP values follow the order water < methanol < gas, which indicates that the stronger the solvent polarity, the higher the electron-donating ability of the molecule.71
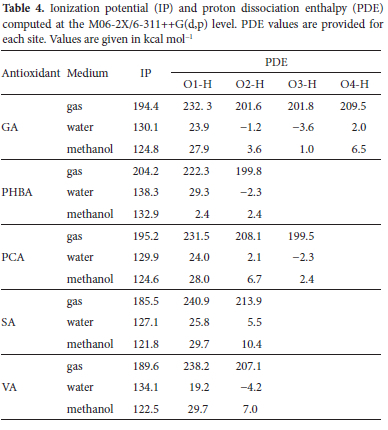
The proton dissociation enthalpy (PDE) characterizes the second step of the SET-PT mechanism and serves to decipher the thermodynamically preferred OH group for deprotonation from the cation radical. It can be seen from Table 4 that the lowest PDE in all the three environments is associated with the OH group in the para position and agrees well with the results based on BDEs. Moreover, PCA has the lowest PDE in the gas phase, while this applies to GA in polar solution. Like IP values, there are substantial decreases of PDEs in solution as compared to the gas phase, which may be ascribed to the high stabilization of the positively charged proton in polar solution. Note that, in the gas phase, the lowest PDEs suggest the following order of antioxidant activity SA < VA < GA < PHBA < PCA, which is slightly different from the one obtained in solution, i.e SA < VA < PHBA < PCA < GA. The negative PDE values predicted for some OH groups may suggest a plausible mediation of the solvent in the heterolytic breakage of the O-H bonds of the radical cation, pulling and favoring the ejection of the proton with no need for extra energy. Wang et al. 72 reported similar data in their investigation of the antioxidant activity of genistein and its nitro and amino derivatives.
SPLET mechanism
Table 5 presents proton affinities (PAs) and electron transfer enthalpies (ETEs) ofGA, PHBA, PCA, SA, and VA in the gas phase and polar (H2O and MeOH) solutions at the M06-2X/6-311++G(d,p) level. In the gas phase, PA values are comprised between 329.2 and 344.0 kcal mol-1, the lowest and highest boundaries being associated with the O2-H and O4-H groups of GA. It must be noted that in the gas phase, the O2-H bond has the lowest PA and should be the most likely to undergo a heterolytic breakage. This group corresponds to the para position with the respect to the carboxylic group, except for GA (meta). In polar solutions, this applies to the O1-H bond for all the molecules, except for GA in water where O4-H has the lowest PA of 30.5 kcal mol-1. Inspection of Table 5 reveals that PAs drop considerably when going from the gas phase to polar solution (where PA values are lower than 39.1 kcal mol-1), following the order gas-phase << water < methanol.
This pattern can be related to the high stability of the radical anion and proton in solution as compared to the gas phase. Considering the lowest PA values in the gas phase, one can deduce the following order of antioxidant activity PHBA~SA <VA<PCA<GA in the gas phase, which changes to PHBA ~ VA < PCA < SA < GA in the polar medium.
After proton dissociation, the next step of the SPLET mechanism consists of the transfer of an electron from ArO - to the free radical. The capability to release this electron density, which is measured by the electron transfer enthalpy (ETE), tells how easily the anionic form of the antioxidant completes this step. ETE values listed in Table 4 are between 58.0 and 89.4 kcal mol-1 in the gas phase and 91.1 and 136.8 kcal mol-1 in polar solution. In the gas phase, site O2 has the lowest ETE value, except in the case of GA for which it is site O4 instead. As such, one can expect these sites (O2 for all, except GA) to be enriched in extra electron density by a striking free radical. Moreover, looking at the lowest ETE values in the gas phase, the anionic form of VA appears to be the ablest to trap an electron from a free radical, followed respectively by that of GA, PCA, SA, and PHBA.
As opposed to PAs, ETE values increase in solution, but their increase is less pronounced than the one associated to the decrease of PA values. Thus, the overall effect is a decrease in the energy needed to pass the second step as compared to the gas phase. This finding can be explained by the high solubility of the radical anion (ArO-) in polar solvents. Let us note that site O2 has the lowest ETE in both solvents for all compounds except GA, for which site O3 and O4 come up with the lowest ETE values of 91.1 and 93.6 kcal mol-1 respectively in water and methanol. In line with the lowest ETE values, the electron transfer ability of these compounds is as follows: PHBA < SA < PCA < VA ~ GA in both water and methanol and is very close to that observed in the gas phase.
Thermodynamically preferred mechanism
To figure out the preferred mechanistic pathway of antioxidant activity, several researchers have relied on the comparison of thermodynamics parameters characterizing the first step of each mechanism.44,73 The same approach is here applied. Recall that IP and PA values describe the first step of the SET-PT and SPLET mechanisms, while BDEs describe the unique reaction of the HAT mechanism. Looking at Tables 2, 4-5, we notice that BDEs are substantially smaller than IPs and PAs in the gas phase. This suggests that the antioxidant activity of GA, PHBA, PCA, SA, and VA is dictated by the HAT mechanism in the gas phase and corroborates with Urbaniak et al.74 As such, PCA and PHBA should display respectively the highest and lowest antioxidant activity in the gas phase. Furthermore, Tables 2, 4-5 reveal that, in polar solution, PAs are very low as compared to BDEs and IPs. This finding confirms the SPLET mechanism as being the most favorable in water and methanol. Therefore, GA and PHBA are respectively the most and least active in the polar medium. Note that, regardless of the environment, PHBA seems to be the least active of the set.
Reactivity
Frontier molecular orbitais
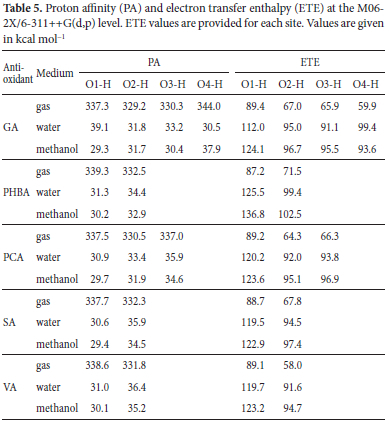
Frontier molecular orbitals (FMOs) are widely used to probe the reactivity of molecular systems.75 Most often, one can rely on the electronic density distribution in these orbitals to predict the most preferred attack site for free radicals and other reactive agents.73 More interestingly, previous studies have shown that the free radical scavenging ability of phenolic compounds can be related to the energy of the HOMO such that molecules with higher HOMO energies are likely to be more active due to the stronger electron-donating ability.44,73 To check this "HOMO rule" and assess the local reactivity of the compounds considered here, FMOs of the five antioxidants were computed in the three environments. Figure 3 displays the electron density distribution of the FMOs of our antioxidants in the gas phase, water and metanol, as well as the corresponding energies.
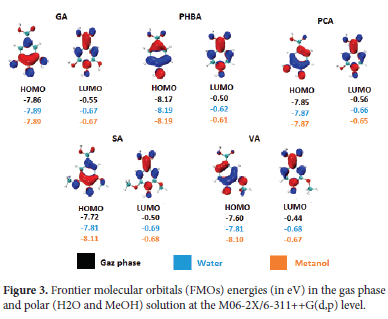
Figure 3 shows that the electron densities of the HOMO and LUMO are delocalized over a big portion of these molecules. The concentration of this density on O atoms and C atoms of the benzene ring suggests these atoms to be the preferred attack site for free radicals.72 However, the reactions on C and C=O sites have not been considered and may constitute the core of a separate study. From Table 6, HOMO energies are comprised between -7.60 and -7.86 eV in the gas phase and range from -7.81 to -8.19 eV in polar solutions. In the gas phase, VA and PHBA have respectively the highest and lowest HOMO energies estimated at -7.60 and -8.17 eV. Assuming the validity of the "HOMO rule", VA should be the strongest electron donor in the gas phase and PHBA the weakest. Applying the same reasoning in the polar phase leads to postulate SA as the best electron donor with the highest HOMO energy of -7.81 eV. HOMO energies listed in Figure 3 suggest the following orders of free electron donor ability: PHBA < GA ~ PCA < SA < VA and PHBA < VA < GA < PCA < SA in gas and polar phases respectively, which do not corroborate with any of the patterns of antioxidant activity predicted based on the various thermodynamic descriptors, i.e., BDE, IP, PDE, PA, and ETE values. This observation is paradoxical and may indicate that the HOMO rule is not absolute and may be misleading in the discussion of the antioxidant activity of some molecules. It is then advised to be cautious with it.
Fukui function f0
As demonstrated earlier, the HAT mechanism dictates the antioxidant activity of these molecules in the gas phase. Recall the HAT is concerned with the abstraction of an H atom by a reactive free radical. Therefore, it can be expected that the most reactive site towards free radicals will result in lower BDE and higher activity. To elucidate this question, we have computed the fukui function f 0 of all the neutral molecules in the gas phase. Figure 4 shows isosurfaces of the fukui f0 function, as well as condensed values on atoms estimated using NBO charges. Let it be noted that other population analyses could have been used to obtain atomic charges, the Hirshfeld protocol being among the most favorite.76,77 Figure 4 displays 0.003 isosurfaces of f0 as well as condensed values on O atoms involved in O-H bonds.
It is clear from Figure 4 that positive values of f0 are concentrated on O atoms, as well as on the C atoms in alpha and para-position with the respect to the carboxylic group. As aforementioned, only H abstractions at hydroxylic O-H groups are considered here. More interestingly, it appears that the carboxylic O atom has the lowest f0 value, while the highest is identified with the one in the para position. This observation corroborates with the local spin densities presented earlier. However, it must be highlighted that both f0 and local spin densities should not be generalized to all five systems. In other words, their values help in understanding the local reactivity of a given molecule but must not be used to compare local reactivities of different molecules.
CONCLUSION
The antioxidant activity of five benzoic acid derivatives has been investigated following three possible mechanisms of antioxidant activity in gas and polar phases. Our findings suggest that the hydrogen atom transfer (HAT) is the preferred mechanistic pathway in the gas phase, while the sequential proton loss electron transfer (SPLET) is favored in polar mediums. Protocatechuic and gallic acids (PCA and GA) are the most active in gas and polar solutions respectively, whereas PHBA is the least active in all the environments considered. Solvents are found to induce considerable changes in the enthalpies of charged species, explaining some drastic variations in proton affinities (PA) and Proton Dissociation Enthalpies (PDE) when going from the gas phase to polar solutions. The O-H group in the para position of the carboxylic group (O3-H for GA and O2-H for the rest) is the most reactive site in the gas phase, while in solution it is either the O1-H site for PHBA, PCA, SA, and VA, or O4-H for GA. The Fukui function f 0 and local spin densities seem to be good indicators of the local reactivity of these molecules in the gas phase. Finally, we have shown that H abstraction radicals gain in stability as they lose in structural aromaticity.
ACKNOWLEDGMENTS
Bienfait K. Isamura is grateful to Prof. Kevin Lobb at Rhodes University and the Center for High Performance Computing (CHPC) for having provided the resources used to carry out this study (project CHEM0802). He is also thankful to the BEBUC scholarship program for financial support.
DISCLOSURE STATEMENT
The authors declare no conflict of interest
SUPPLEMENTARY MATERIAL
The supplementary information contains optimized geometries of the closed-shell antioxidants and the associated H-abstraction radicals in vacuum, water, and methanol environments as well as gas-phase HOMA values of the benzene ring before and after removing one H atom.
ORCID IDs
Bienfait K. Isamura: https://orcid.org/0000-0003-3278-3284
Issofa Patouossa: https://orcid.org/0000-0001-6761-9295
Isaac E. Kaba: https://orcid.org/0000-0003-0234-0626
Aristote Matondo: https://orcid.org/0000-0002-6246-9803
Pius T. Mpiana: https://orcid.org/0000-0002-8590-6017
REFERENCES
1. Elaka IK, Kapepula PM, Ngombe NK, Mpoyi JM, Lukusa DM, Muabilwa MM, Gabha'Bey CK, Lukukula CM, Lukebakio BN, Okuna MK, Kialengila DM, Kantola PT. Microscopic features, antioxidant and antibacterial capacities of plants of the congolese cosmetopoeia, raw materials of cosmeceuticals. J Biosci Med. 2020;8(9):149-166. https://doi.org/10.4236/jbm.2020.89013 [ Links ]
2. Pruteanu LL, Bailey DS, Gràdinaru AC, Jântschi L. The Biochemistry and Effectiveness of Antioxidants in Food, Fruits, and Marine Algae. Antioxidants. 2023; 12(4):860. https://doi.org/10.3390/antiox12040860 [ Links ]
3. Patrick L. Mercury toxicity and antioxidants: Part I: role of glutathione and alpha-lipoic acid in the treatment of mercury toxicity. Altern Med Rev. 2002;7(6):456-471. [ Links ]
4. Matsumura Y, Kitabatake M, Kayano S-i, Ito T. Dietary Phenolic Compounds: Their Health Benefits and Association with the Gut Microbiota. Antioxidants. 2023; 12(4):880. https://doi.org/10.3390/antiox12040880 [ Links ]
5. Llano S, Gómez S, Londono J, Restrepo A. Antioxidant activity of curcuminoids. Phys Chem Chem Phys. 2019:21:3752-3760. https://doi.org/10.1039/C8CP06708B [ Links ]
6. Kâhkònen MP, Heinonen M. Antioxidant activity of anthocyanins and their aglycons. J Agric Food Chem. 2003;51(3):628-633. https://doi.org/10.1021/jf025551i [ Links ]
7. Xu DP, Li Y, Meng X, Zhou T, Zhou Y, Zheng J, Zhang JJ, Li HB. Natural Antioxidants in Foods and Medicinal Plants: Extraction, Assessment and Resources. Int J Mol Sci. 2017;18(1):96. https://doi:10.3390/ijms18010096 [ Links ]
8. Krinsky NI. Carotenoids as antioxidants. Nutrition. 2001;17(10):815-817. https://doi.org/10.1016/s0899-9007(01)00651-7 [ Links ]
9. Corino C, Rossi R. Antioxidants in Animal Nutrition. Antioxidants (Basel). 2021;10(12):1877 https://doi:10.3390/antiox10121877 [ Links ]
10. Regenstein JM, Zhou P. Collagen and gelatin from marine by-products. In Maximising the Value of Marine By-Products( Shahidi F ed). Woodhead Publishing: Shaxton, UK. 2007: 279-303. [ Links ]
11. Konuray G, Erginkaya Z. Antimicrobial and antioxidant properties of pigments synthesized from microorganisms, in Battle against microbial pathogens: Basic science, technological advances and educational Programs (Méndez-Vilas A ed). Formatex Research Center: Badajoz, Spain. 2015:2733. [ Links ]
12. Ren CY, Wu EL, Hartmann EM, Zhao HP. Biological mitigation of antibiotic resistance gene dissemination by antioxidant-producing microorganisms in activated sludge systems. Environ Sci Technol. 2021;55(23):15831-15842. https://doi.org/10.1021/acs.est.1c04641 [ Links ]
13. Shahidi F, Zhong Y. Measurement of antioxidant activity. J Funct Foods. 2015,18: 757-781. https://doi.org/10.1016/j.jff.2015.01.047 [ Links ]
14. Torres de Pinedo A, Penalver P, Morales JC. Synthesis and evaluation of new phenolic-based antioxidants: Structure-activity relationship. Food Chem. 2007; 103(1):55-61. https://doi.org/10.1016/j.foodchem.2006.07.026 [ Links ]
15. Dorothée DT, Philippe BB, Damase NV, Christian MT, Damien ST Koto-te-Nyiwa N , Philippe VT, Pius TM. Chemo-type of essential oil of ocimum basilicum L from DR Congo and relative in vitro antioxidant potential to the polarity of crude extracts. Asian Pac J Trop Biomed. 2016;6:1022-1028. https://doi.org/10.1016/j.apjtb.2016.08.013 [ Links ]
16. Pietta PG. Flavonoids as antioxidants. J Nat Prod. 2000;63(7): 1035-1042. https://doi.org/10.1021/np9904509 [ Links ]
17. Scalbert A, Johnson IT, Saltmarsh M. Polyphenols: antioxidants and beyond. Am J Clin Nutr. 2005;81(1): 215-217. https://doi.org/10.1093/ajcn/81.1.215S [ Links ]
18. Pandey KB, Rizvi SI. Plant polyphenols as dietary antioxidants in human health and disease. Oxid Med Cell Longev. 2009;2(5):270-8. https://doi.org/10.4161/oxim.2.5.9498 [ Links ]
19. Velika B, Kron I. Antioxidant properties of benzoic acid derivatives against superoxide radical. Free Rad and Antiox. 2012;2(4):62-67. https://doi.org/10.5530/ax.2012.4.11 [ Links ]
20. Al-Haidari RA, Al-Oqail MM. New benzoic acid derivatives from Cassia italica growing in Saudi Arabia and their antioxidant activity. Saudi Pharm J. 2020;28(9):1112-1117. d https://doi.org/10.1016Ai.jsps.2020.07.012 [ Links ]
21. Saravanan N, Rajasankar S, Nalini N. Antioxidant effect of 2-hydroxy-4-methoxy benzoic acid on ethanol-induced hepatotoxicity in rats. J Pharm Pharmacol. 2007;59(3):445-53. https://doi.org/10.1211/jpp.593.0015 [ Links ]
22. Skorna P, Michalík M, Klein E. Gallic acid: thermodynamics of the Homolytic and Heterolytic phenolic O-H Bonds Splitting-off. Acta Chim Slovaca. 2016;9: 114-123. https://doi.org/10.1515/acs-2016-0020 [ Links ]
23. Mohajeri A, Asemani SS. Theoretical investigation on antioxidant activity of vitamins and phenolic acids for designing a novel Antioxidant. J Mol Struct. 2009;930(1-3):15-20. https://doi.org/10.1016/j.molstruc.2009.04.031 [ Links ]
24. Kalita D, Kar R, Handique JG. A theoretical study on the antioxidant property of gallic acid and its derivatives. J Theor Comput Chem. 2012;11(2):391-402. https://doi.org/10.1142/S0219633612500277 [ Links ]
25. Shang Y, Zhou H, Li X, Zhou J, Chen K. Theoretical studies on the antioxidant activity of viniferifuran. New J Chem. 2019;43: 15736-15742. https://doi.org/10.1039/C9NJ02735A [ Links ]
26. Yan-Zhen Z, Geng D, Da-Fu C, Qin L, Rui G, Zhong-Min F. Theoretical studies on the antioxidant activity of pinobanksin and its ester derivatives: Effects of the chain length and solvent. Food Chem.2018;240: 323-329. https://doi.org/10.1016/j.foodchem.2017.07.133 [ Links ]
27. Alisi IO, Uzairun A, Abechi SE. Molecular design of curcumin analogues with potent antioxidant properties and thermodynamic evaluation of their mechanism of free radical scavenge. Bull Natl Res Cent. 2020;44: 137. https://doi.org/10.1186/s42269-020-00391-z [ Links ]
28. Vásquez-Espinal A, Yanez O, Osorio E, Carlos A, Garcia-eltran O, Lina MR, Bruce KC, William T. Theoretical study of the antioxidant activity of quercetin oxidation products. Front Chem. 2019;7:1-10. https://doi.org/10.3389/fchem.2019.00818 [ Links ]
29. Dai J, Mumper RJ. Plant phenolics: extraction, analysis and their antioxidant and Anticancer properties. Molecules. 2010;15(10):7313-7352. https://doi.org/10.3390/molecules15107313 [ Links ]
30. Liu Q, Luo L, Zheng L. Lignins: biosynthesis and biological functions in plants. Int J Mol Sci. 2018;19(2):335. https://doi.org/10.3390/ijms19020335 [ Links ]
31. Manuja R, Sachdeva S, Jain A, Jasmine C. A comprehensive review on biological activities of p-hydroxy benzoic acid and its derivatives. Int J Pharm Sci Rev Res. 2013;22(2):109-115. [ Links ]
32. Cikman O, Soylemez O, Ozkan OF, Kiraz HA, Sayar I, Ademoglu S, Taysi S, Karaayvaz M. Antioxidant activity of syringic acid prevents oxidative stress in i-arginine-induced acute pancreatitis: An experimental study on rats. Int Surg. 2015;100(5):891-6. https://doi.org/10.9738/INTSURG-D-14-00170.1 [ Links ]
33. Tai A, Sawano T, Ito H. Antioxidative properties of vanillic acid esters in multiple antioxidant assays. Biosci Biotechnol Biochem. 2012;76(2):314-8. https://doi.org/10.1271/bbb.110700 [ Links ]
34. Verma S, Singh A, Mishra A. Gallic acid: molecular rival of cancer. Environ Toxicol Pharmacol. 2013;35(3):473-85. https://doi.org/10.1016/j.etap.2013.02.011 [ Links ]
35. Vinkovic VI, Samobor V, Bojic M, Medic-Saric M, Vukobratovic M, Erhatic R, Horvat D, Matotan Z. The effect of grafting on the antioxydating properties of tomato (Solanum lycopersicum L.). Spanish J Agric Res. 2011;9(3):844-851. https://doi.org/10.5424/sjar/20110903-414-10 [ Links ]
36. Ma J, Luo XD, Protiva P, Yang H, Ma C, Basile MJ, Weinstein IB, Kennelly EJ. Bioactive novel polyphenols from the fruit of Manilkara zapota (Sapodilla). J Nat Prod. 2003;66(7):983-9866. https://doi.org/10.1021/np020576x [ Links ]
37. Shahrzad S, Bitsch I. Determination of some pharmacologically active phenolic acids in juices by high-performance liquid chromatography. J Chromatogr A. 1996 16;741(2):223-31. https://doi.org/10.1016/0021-9673(96)00169-0 [ Links ]
38. Eyles A, Davies NW, Mitsunaga T, Mihara R, Mohammed C. Role of Eucalyptus globulus wound wood extractives: evidence of superoxide dismutase-like activity. For Path. 2004;34(4):225-232. http://doi.org/10.1111/j.1439-0329.2004.00361.x [ Links ]
39. Murugananthan M, Raju GB, Prabhakar S. Removal of Tannins and polyhydroxy phenols by electro-chemical. J Chem Technol Biotechnol. 2005;80(10):1188-1197. https://doi.org/10.1002/jctb.1314 [ Links ]
40. Arunkumar S, Ilango K, Manikandan RS, Ramalakshmi N. Synthesis and anti-inflammatory activity of some novel pyrazole derivatives of gallic acid, E-J Chem. 2009;6:123-129. https://doi.org/10.1155/2009/128586 [ Links ]
41. Ow YY, Stupans I. Gallic acid and gallic acid derivatives: effects on drug metabolizing enzymes. Curr Drug Metab. 2003;4(3):241-248. https://doi.org/10.2174/1389200033489479 [ Links ]
42. Mirvish SS, Cardesa A, Wallcave L, Shubik P. Induction of mouse lung adenomas by amines or ureas plus nitrite and by N-nitroso compounds: effect of ascorbate, gallic acid, thiocyanate, and caffeine. J Natl Cancer Inst. 1975;55(3):633-636. https://doi.org/10.1093/jnci/55.3.633 [ Links ]
43. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09 Gaussian Inc. Wallingford CT. 2009. [ Links ]
44. Zheng YZ, Deng G, Liang Q, Chen DF, Guo R, Lai RC. Antioxidant activity of quercetin and its glucosides from propolis: A theoretical study. Sci Rep. 2017 8;7(1):7543. https://doi.org/10.1038/s41598-017-08024-8 [ Links ]
45. Muthukumaran J, Srinivasan S, Venkatesan RS, Ramachandran V, Muruganathan U. Syringic acid, a novel natural phenolic acid, normalizes hyperglycemia with special reference toglycoprotein components in experimental diabetic rats. J Acu Dis. 2013;2(4):304-309. https://doi.org/10.1016/S2221-6189(13)60149-3 [ Links ]
46. Rimarcík J, Lukes V, Klein E, Michal I. Study of the solvent effect on the enthalpies of homolytic and heterolytic N-H bond cleavage in p-phenylenediamine and tetracyano-p-phenylenediamine. J Mol Struct: Theochem. 2010;952(1-3):25-30. https://doi.org/10.1016/j.theochem.2010.04.002 [ Links ]
47. Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models. Chem Rev. 2005;105(8):2999-3093. https://doi.org/10.1021/cr9904009 [ Links ]
48. Tomasi J, Mennucci B, Cances E. The IEF version of the PCM solvation method : an overview of a new method addressed to study molecular solutes at the QM ab initio level. J Mol Struct: Theochem. 1999;464(1-33):211-226. https://doi.org/10.1016/S0166-1280(98)00553-3 [ Links ]
49. Cramer CJ, Truhlar DG. Implicit solvation models: equilibria, structure, spectra, and dynamics. Chem Rev. 1999;99(8):2161-2200. https://doi.org/10.1021/cr960149m. [ Links ]
50. Kruszewski J, Krygowski TM. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972;13(36):3839-3842. https://doi.org/10.1016/S0040-4039(01)94175-9 [ Links ]
51. Lee C, Yang W, Parr RG. Local softness and chemical reactivity in the molecules CO, SCN- and H2CO. J Mol Struct: Theochem. 1955;163:305-313. https://doi.org/10.1016/0166-1280(88)80397-X [ Links ]
52. Martinez C, Rivera JL, Herrera R, Rico JL, Flores N, Rutiaga JG, López P. Evaluation of the chemical reactivity in lignin precursors using the Fukui function. J Mol Model. 2008;14(2):77-81. https://doi.org/10.1007/s00894-007-0253-0 [ Links ]
53. Ma Y, Liang J, Zhao D, Yue-Lei C, Shen J, Bing X. Condensed Fukui function predicts innate C-H radical functionalization sites on multi-nitrogen containing fused arenes. Rsc. Adv. 2014;4:17262-17264. https://doi.org/10.1039/C4RA01853B [ Links ]
54. Hanson RM. Jmol-a paradigm shift in crystallographic visualization. J. Appl. Crystallogr. 2010;43(5):1250-1260. http://doi.org/10.1107/S0021889810030256/kk5066sup24.zip [ Links ]
55. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33-38. https://doi.org/10.1016/0263-7855(96)00018-5 [ Links ]
56. Lu T, Chen F. Multiwfn: A multifunctional wavefunction analyzer. J Comput Chemy. 2012;33: 80-92. http://doi.org/10.1002/jcc.22885 [ Links ]
57. Glendening ED, Landis CR, Weinhold F. NBO 6.0: natural bond orbital analysis program. J Comput Chem. 2013;34(16):1429-1437. http://doi.org/10.1002/jcc.23266. [ Links ]
58. Siquet C, Paiva-Martins F, Lima JL, Reis S, Borges F. Antioxidant profile of dihydroxy- and trihydroxyphenolic acids-a structure-activity relationship study. Free Radic Res. 2006;40(4):433-442. http://doi.org/10.1080/10715760500540442 [ Links ]
59. Gordon MS, Jensen JH. Understanding the hydrogen bond using quantum chemistry. Acc Chem Res. 1996;29(11):536-543. http://doi.org/10.1021/ar9600594 [ Links ]
60. Mazzone G, Toscano M, Russo N. Density functional predictions of antioxidant activity and UV spectral features of nasutin A, isonasutin, ellagic acid, and one of its possible derivatives. J Agric Food Chem. 2013;61(40):9650-9657. http://doi.org/10.1021/jf403262k [ Links ]
61. Hamadouche S, Ounissi A, Baira K, Ouddai N, Balsamo M, Erto A, Benguerban Y. Theoretical evaluation of the antioxidant activity of some stilbenes using the density functional theory. J Mol Struct. 2021;1229: 29496. https://doi.org/10.1016/j.molstruc.2020.129496 [ Links ]
62. Evgeny D, Taisa D. Dissociation energies of O - H bonds of phenols and hydroperoxides, application of thermodynamics to biological and materials science. InTech. 2010;57:1858-1866. https://doi.org/10.5772/13290 [ Links ]
63. Alberti A, Amorati R, Campredon M, Lucarini M, Macciantelli M. Antioxidant activity of some simple phenols present in olive oil. Acta Aliment.2009;38(4):427-436. https://doi.org/10.1556/AAlim.38.2009.4.3 [ Links ]
64. Mazzone G, Malaj N, Russo N, Toscano M. Density functional study of the antioxidant activity of some recently synthesized resveratrol analogues. Food Chem. 2013;141(3):2017-024. https://doi.org/10.1016/j.foodchem.2013.05.071 [ Links ]
65. Leopoldini M, Chiodo SG, Russo N, Toscano M. Detailed investigation of the OH radical quenching by natural antioxidant caffeic acid studied by quantum mechanical models. J Chem Theory Comput. 2011;7(12):4218-233. https://doi.org/10.1021/ct200572p [ Links ]
66. Leopoldini M, Marino T, Russo N, Toscan M, Antioxidant properties of phenolic compounds: H-atom versus electron tansfer mechanism. J Phys Chem A. 2004;108(22):4916-4922. https://doi.org/10.1021/jp037247d [ Links ]
67. Lin L, Zhu J. Antiaromaticity-promoted radical stability in a-methyl heterocyclics. J Org Chem. 2021;86(21):15558-15567. https://doi.org/10.1021/acs.joc.1c02050 [ Links ]
68. Ma Y, Feng Y, Diao T, Zeng W, Zuo Y. Experimental and theoretical study on antioxidant activity of the four anthocyanins. J Mol Struct. 2020;1024:127509. https://doi.org/10.1016/j.molstruc.2019.127509 [ Links ]
69. Wright JS, Johnson ER, DiLabio GA. Predicting the activity of phenolic antioxidants: theoretical method, analysis of substituent effects, and application to major families of antioxidants. J Am Chem Soc. 2001;123(6):1173-1183. https://doi.org/10.1021/ja002455u [ Links ]
70. Fifen JJ, Nsangou M, Dhaouadi Z, Motapon O, Solvent effects on the antioxidant activity of 3,4-dihydroxyphenylpyruvic acid: DFT and TD-DFT studies. Comput Theor Chem, 2011;966(1-3):232-243. https://doi.org/10.1016/j.comptc.2011.03.006 [ Links ]
71. Chen J, Yang J, Ma L, Li J, Shahzad N, Kim CK. Structure-antioxidant activity relationship of methoxy, phenolic hydroxyl, and carboxylic acid groups of phenolic acids. Sci Rep. 2020;10(1):2611. https://doi.org/10.1038/s41598-020-59451-z [ Links ]
72. Wang G, Xue Y, An L, Youguang Z, Yunyan D, Ling Z, Yi L, Theoretical study on the structural and antioxidant properties of some recently synthesised 2,4,5-trimethoxy Chalcones. Food Chem. 2015;171: 89-97. https://doi.org/10.1016/j.foodchem.2014.08.106 [ Links ]
73. Xue Y, Zheng Y, An L, Dou Y, Liu Y. Density functional theory study of the structure-antioxidant activity of polyphenolic deoxybenzoins. Food Chem. 2014;151:198-206. https://doi.org/10.1016/j.foodchem.2013.11.064 [ Links ]
74. Urbaniak A, Szelag M, Molski M. Theoretical investigation of stereochemistry and solvent influence on antioxidant activity of ferulic acid. Comput Theor Chem. 2013;1012: 33-40. https://doi.org/10.1016/J.COMPTC.2013.02.018 [ Links ]
75. Ali A, Khalid M, Abid S, Tahir MN, Iqbal J, Ashfaq M, Kanwal F, Lu C, Rehman MFu. Green synthesis, SC-XRD, non-covalent interactive potential and electronic communication via DFT exploration of pyridine-based hydrazone. Crystals. 2020; 10(9):778. https://doi.org/10.3390/cryst10090778 [ Links ]
76. Hirshfeld FL. Bonded-atom fragments for describing molecular charge densities. Theoret. Chim. Acta. 1977;44:129-138. https://doi.org/10.1007/BF00549096 [ Links ]
77. Isamura BK, Patouossa I, Muya JT, Lobb KA. Unveiling the reactivity of truxillic and truxinic acids (TXAs): deprotonation, anion... H-O, cation... O and cation... n interactions in TXA0... Y+ and TXA0... Z- complexes (Y= Li, Na, K; Z= F, Cl, Br), Struct Chem. 2022;34(1):97-112. https://doi.org/10.1007/s11224-022-01965-5 [ Links ]
Received 16 July 2022
Revised 26 March 2023
Accepted 26 May 2023
* To whom correspondence should be addressed: Email: isamurabft@gmail.com

