



![C-H activation: a Critical Evaluation of a Published Method and its Application Towards Inherently Chiral Calix[4]arenes](/img/pt/prev.gif)






Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Chemistry
versão On-line ISSN 1996-840X
versão impressa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.77 Durban 2023
http://dx.doi.org/10.17159/0379-4350/2023/v77a04
RESEARCH ARTICLE
Re-screening and preliminary in vitro evaluation of resorcinol based Hsp90 inhibitors for potential protocidal activity
Théoneste UmumararunguI, II, *; Michelle IsaacsIII; Heinrich C. HoppeIII, IV; Eleonora D. GoosenII; Setshaba D. KhanyeII, III, *
IDepartment of Pharmacy, School of Medicine and Pharmacy, College of Medicine and Health Sciences, University of Rwanda, Rwanda
IIDivision of Pharmaceutical Chemistry, Faculty of Pharmacy, Rhodes University, Makhanda, South Africa
IIICentre for Chemico- and Biomedicinal Research, Rhodes University, Makhanda, South Africa
IVDepartment of Biochemistry and Microbiology, Rhodes University, Makhanda, South Africa
ABSTRACT
This study explored a series of resorcinol derivatives originally identified as heat shock protein 90 inhibitors for activity against protozoan parasites. The repurposing of this series revealed that some of these compounds are active against chloroquine-sensitive Plasmodium falciparum (3D7) and Trypanosoma brucei brucei strains with IC50 values below 10 μM without obvious cytotoxic effects against human cervix adenocarcinoma (HeLa) cells at 25 μM.
Keywords: drug-likeness, Plasmodium falciparum, resorcinol derivatives, sleeping sickness, Trypanosoma brucei brucei,
INTRODUCTION
Malaria is still considered a serious global concern to a large population in tropical regions. In 2020, WHO reported approximately 627 000 malaria deaths, with sub-Saharan Africa accounting for 96% of these.1 The slight increase in deaths in 2020 from 558 000 in 2019 is attributable to the COVID-19 pandemic, which stalled malaria control programmes. Although a slight upward trend emerged in 2020, global efforts in the last two decades have led to a significant decline in deaths among children under five years in sub-Saharan Africa.1 Following a period in which antimalarial drugs lost efficacy due to drug resistance, the World Health Organization (WHO) declared artemisininbased combination therapies (ACTs) the first line of defence against uncomplicated P. falciparum malaria.2 Concerns of emergence of clinical resistance to ACTs - including to both artemisinin derivatives and partner drugs such as the 4-aminoquinolines or aminoalcohols - are on the rise.3 Thus, fears that this resistance will spread to resource-limited endemic parts of Africa and reverse the gains achieved against malaria eradication are justified.3-5 Despite numerous on-going efforts to bring a malaria vaccine to the clinic, developing an effective vaccine suitable for a diverse population remains an elusive target.4,6 More recently, clinical trials for the novel, promising fast-acting anti-malarial drug candidates MMV048, P218 and DSM265 (Figure 1) were discontinued because of clinical toxicity.7 Such discontinuation of clinical trials for investigational new drugs (INDs) is not restricted to malaria drug discovery and development. Rather, it highlights the high attrition rate of compounds in drug development endeavours prevalent across all drug discovery projects. The limitations of existing antiparasitic drugs in clinical use, coupled with the high attrition rates encountered in drug development phases, thus obviates the need to continue searching for new classes of compounds with novel mechanisms of action.3,4On the other hand, human African Trypanosomiasis (HAT) or sleeping sickness, caused by Trypanosoma brucei parasites is an important insect-borne protozoan parasitic disease that affects poor populations in remote areas of sub-Saharan Africa.8,9 Geographically, T. b. gambiense is common in central and West Africa, while T. b. rhodesiense is widespread in the Eastern part of Africa.10-12 These parasites are transmitted to humans following bites by tsetse flies during feeding on human blood. The treatment options for HAT include a handful of FDA-approved drugs: pentamidine, suramin, melarsoprol, eflornithine and nifurtimox, which have been used for several decades.13,14 These drugs, however, have shortcomings that limit their clinical use, such as reduced efficacy, toxic side effects and the emergence of resistance. Currently, the first-line treatment includes pentamidine for stage 1 of the disease. On other hand, nifurtimox-eflornithine combination therapy (NECT) is used to treat stage 2 of the disease, an important stage of the disease wherein parasites spread and invade the central nervous system (CNS). The recent increased campaigns for alternative drug candidates have yielded two effective compounds, fexinidazole and SCYX-7158 (Figure 1), which are currently undergoing clinical trials for the treatment of HAT.14-18 Moreover, these drug candidates show positive outcomes for treating stages 1 and 2 of the disease.
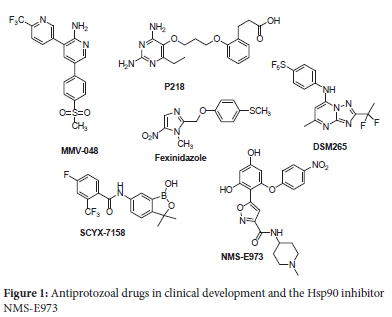
Exploiting inhibitors of human enzymes that are homologous to parasitic equivalents is an efficient strategy that researchers have used to successfully identify new antiparasitic agents.19-22 The heat shock protein 90 (Hsp90) inhibitors, which have drawn considerable attention as anticancer agents, are increasingly being explored as potential antiprotozoal agents.23-25 Hsp90, a molecular chaperone that regulates protein folding and gene expression, is critical for survival of protozoan parasites such as P. falciparum, T. evansi, Eimeria tenella, Leishmania donovani and Toxoplasma gondii under the severe environmental conditions in which they exist within the hosts.24 Previous studies on Hsp90 inhibitors showed that treating mice infected with malaria and surra with geldanamycin - an Hsp90 inhibitor - prolonged their survival rate compared with the untreated ones.26 A recent study by Giannini et al explored Hsp90 inhibitors, originally developed for cancer treatment, for their ability to inhibit growth of human protozoan parasites in vitro.27 Data from this this study revealed that P. falciparum and T. b. rhodesiense parasites showed sensitivity to the Hsp90 inhibitors, while T. cruzi and Giardia lamblia were less sensitive. Considering the effects of these Hsp90 inhibitors against protozoan parasites, we sought to repurpose the resorcinol derivatives previously reported by Brasca et al28 for their potential antiplasmodial and antitrypanosomal activities against P. falciparum and T. b. brucei, respectively. We hypothesized that the hit compound NMS-E973 (Figure 1) - a selective inhibitor of Hsp90 with significant efficacy against human ovarian A2780 cancer cell line - would be active against protozoan parasites that express Hsp90. The aim of this study was, thus, to repurpose novel derivatives of NMS-E973 Hsp90 inhibitors as antiprarasitic agents, and to determine their potential as growth inhibitors of P. falciparum and T.b. brucei. Herein, we report the synthesis and in vitro evaluation of selected resorcinol derivatives for their potential pharmacological activity against strains of the protozoal parasites P. falciparum and T. b. brucei.
EXPERIMENTAL
Commercially available chemicals and reagents used in this project were purchased from Sigma-Aldrich and Merck and were used without further purification unless stated otherwise. The synthesis and characterisation of compounds TU-011 to TU-019 have been reported previously.28 The full characterisation data is provided in the Supplementary Information file.
In vitro antiplasmodial activity assay
The 3D7 strain P. falciparum was cultured in medium consisting of RPMI 1640 containing 25 mM Hepes (Lonza), supplemented with 5% (w/v) Albumax II (ThermoScientific), 22 mM glucose, 0.65 mM hypoxanthine, 0.05 mg/mL gentamicin and 2-4% (v/v) human erythrocytes. Cultures were maintained at 37 °C under an atmosphere of 5% CO2, 5% O2, 90% N2 gas mixture. To measure the antiplasmodial activity, three-fold serial dilutions of each test compound in culture medium were added to parasite culture (adjusted to 2% parasitaemia, 1% haemotocrit) in 96-well plates and incubated at 37 °C for 48 h. Duplicate wells per compound concentration were used. Parasite lactate dehydrogenase (pLDH) enzyme activity in the individual wells was assessed as previously described.29,30
In vitro antitrypanosomal activity assay
Trypanosoma brucei brucei 427 trypomastigotes were cultured in Iscove's Modified Dulbecco's medium (IMDM, Lonza, Basel Switzerland) and supplemented with 10% fetal calf serum, HMI-9 supplement,31,32 hypoxanthine and penicillin/streptomycin at 37 °C in a 5% CO2 incubator. Serial dilutions of each test compound were incubated with the parasites in 96-well plates at 37 °C for 24 h. The residual parasite viability in the wells was determined by adding 20 μL resazurin toxicology reagent (Sigma-Aldrich) and incubating for an additional 24 h. The reduction of resazurin to resorufin by viable parasites was assessed by fluorescence readings (excitation 560 nm, emission 590 nm) in a Spectramax M3 plate reader (Molecular Devices, San Jose, CA, USA). Fluorescence readings were converted to % parasite viability relative to the average readings obtained from untreated control wells. IC50 values were determined by plotting % viability vs. log[compound] and performing non-linear regression using GraphPad Prism (v. 5.02) software.29
In vitro cell cytotoxicity assay
HeLa cells (Cellonex) seeded in 96-well plates were incubated with 25 μM test compounds for 24 h as previously described.33 The cell viability was assessed using a resazurin fluorescence assay.
Computational methods
Virtual screening of the candidate compounds was done using Autodock vina.34 Firstly, the energy of each ligand was minimised with Vegazz. Then the ligand and the target protein (PfHsp90 (PDB code: 3K60) or human Hsp90 (PDB code: 1BYQ)) were prepared with Autodock tools for docking with Autodock vina. Compounds whose estimated maximum length was less than 15 A, were docked in a 25 A χ 25 A χ 25 A box, and those with an estimated maximum length equal to or larger than 15 A, were docked in a 30 A χ 30 A χ 30 A box. The intention was to increase the docking speed, thus minimising the docking time while preserving the accuracy of predictions of the binding modes of the candidate compounds. Then, the ratio between the inhibition constants of the best binding modes of each ligand for human Hsp90 and PfHsp90 was determined and used to decide which compounds would selectively bind to PfHsp90 in the presence of human Hsp90. The approximate dimensions of the ligands were determined using Vegazz. ADMET properties of synthesised compounds were obtained from SwissADME online server35 using the SMILES inputs of each compound generated by ChemDraw software.
RESULTS AND DISCUSSION
Chemistry
Synthesis of this series has been reported previously by Brasca and co-workers.28 In a slight modification of Brasca's methodology, compounds TU-011 to TU-019 were readily obtained in moderate to good yields (Scheme 1). All the synthesised compounds TU-011 to TU-019 were fully characterised by 1H and 13C NMR and mass spectrometry (MS) and information is provided as the electronic supplementary file.
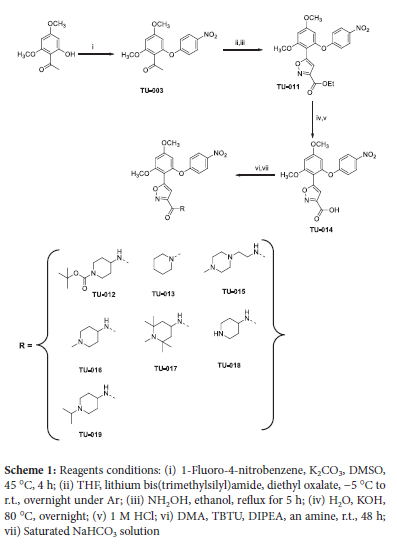
In vitro biological assays
The series of resorcinol derivates TU-014-019 (Scheme 1) were screened in vitro for activity against P. falciparum (3D7 strain) and T. brucei (427 strain). Solutions of compounds TU-014 to TU-019 suspended in DMSO were used to determine their cytotoxicity against human cervix adenocarcinoma (HeLa) cells. Chloroquine, pentamidine and emetine were used as positive controls for antiplasmodial, antitrypanosomal activity and cytotoxicity (Figure S2), respectively. The results are summarised in Table 1 (below) and the electronic supplementary file.
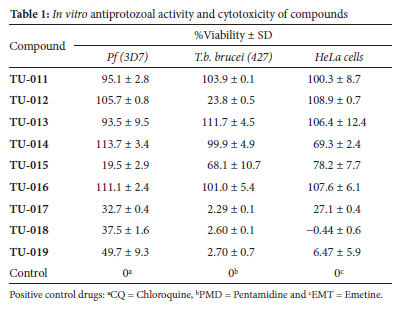
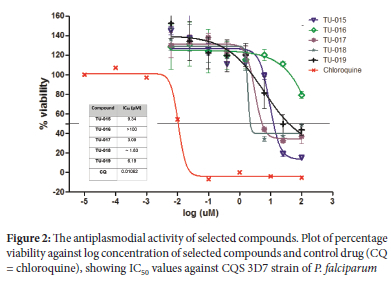
Except for compounds TU-014 (IC50 ~ 25 μΜ), TU-017 (IC50 = 28.5 μΜ), TU-018 (IC50 = 24.8 μΜ) and TU-019 (IC50 ~ 25.0 μΜ), which showed modest cytotoxicity, the rest of compounds showed minimal growth inhibition of HeLa cell line at 100 μΜ concentration, resulting in >70% HeLa cell viability. Initially, the resorcinol series TU-011 to TU-019 was evaluated for in vitro P. falciparum inhibitory activity at a single concentration of 25 μΜ (DMSO) in duplicates using malaria parasite lactate dehydrogenase (pLDH) assays.36 At this concentration, most of the compounds showed no activity, with parasite percentage viability above 80% for most (Table 1). Compounds TU-015 and TU-017 to TU-019 reduced parasite viabilities below the 50% threshold; accordingly, dose-response analyses were conducted
on these compounds to determine the corresponding IC50 values (Figure 2). In general, these compounds were moderately active against the 3D7 strain, with IC50 values below 10 μΜ. Compound TU-018 (IC50 = 1.83 μΜ) emerged as the most active derivate from this series. Similarly, the solutions containing TU-011 to TU-019 were prepared to a single fixed concentration of 25 μg/mL (DMSO). Of the nine compounds, compounds TU-012 and TU-017 to TU-019 suppressed T. b. brucei viability below 30% parasite (Table 1) at 25 μg/mL (DMSO). The IC50 values of the four active compounds were determined, and the data is depicted in Fig. 3. The results show that the compounds were active against T. b. brucei within a narrow IC50 window of 2 - 6 μΜ. Once again, the most active compound was TU-018, with an IC50 value of 2.87 μΜ. The superior activity of TU-018 compared to analogous compounds (e.g., TU-012, TU-016 and TU-019) could be from the increased basicity of the secondary amine of the piperidine ring, which maybe more favourable for accumulation at the site of action and therefore activity against P. falciparum and T.b. brucei, respectively. Compared to the reference drugs choroquine and pentamindine, TU-014 to TU-019 displayed reduced activity.
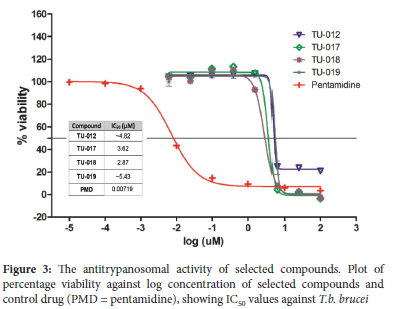
In silico drug-likeness prediction
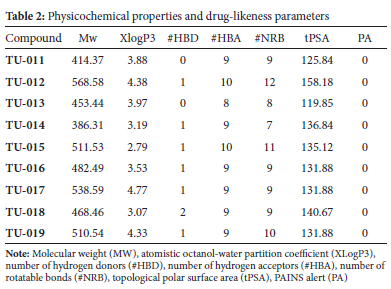
To be effective as a chemotherapeutic agent, a drug must reach a specific target in sufficient concentration. Each year, many promising therapeutic agents that are advanced from early drug discovery stages fail to reach clinical trials because of undesirable drug-likeness and poor pharmacokinetic properties.37,38 Thus, it is critical to profile the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of new molecules intended for medicinal applications. In recent years, researchers have applied open access in silico tools to assess compound ADME parameters in early-phase drug discovery and development processes.35,37 In this work, each compound's drug-likeness was assessed using the SwissADME35 web tool to generate relevant physicochemical parameters with structural inputs derived from the synthesised compounds. The compounds TU-011 to TU-019 were profiled by applying Lipinski's rule of five (Ro5), a decision guide that assesses the theoretical oral bioavailability of orally administered drugs. Lipinski's Ro5 proposes that in order to exhibit good oral bioavailability a compound should have a partition coefficient (XLogP) < 5.0, molecular weight < 500, hydrogen bond acceptors < 10, and donors' < 5. A compound that violates more than one of these rules is likely to demonstrate/have unfavourable oral bioavailability.39 Data from in silico screening of our compound set using SwissADME web tool are shown in Table 2. These in silico ADME data suggest that the compounds have acceptable ADME properties despite compounds TU-012, TU-015, TU-017 and TU-019 showing molecular weights > 500 Da. Within the context of possessing desirable in silico ADME properties, we predict TU-014 to TU-019 to exhibit a low risk of acting as pan-assay interference (PAINS), suggesting they are not promiscuous binders or inhibitors.
Molecular docking studies
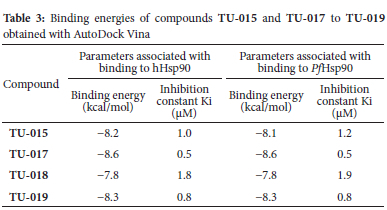
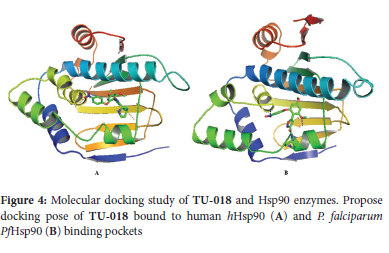
To investigate the possible molecular mechanism responsible for the antiplasmodial activity of selected compounds TU-015 and TU-017 to TU-019, we performed molecular docking analyses using AutoDock Vina with P falciparum Hsp90 (PfHsp90). These compounds showed activity against P. falciparum and T.b. brucei protozoan parasites with IC50 values below 10 μM. We reasoned that such docking analyses would provide the insight into key interactions between ligands TU-015 and TU-017 to TU-019 and PfHsp90 enzyme. The structure of the N-terminal nucleotide, which is the drug binding domain of PfHsp90 has been reported in literature.40 Thus, the ADP-bound crystal structures of human Hsp90 (PDB code: 1BYQ) and PfHsp90 (pDB code: 3K60) were retrieved from the protein data bank to perform the molecular docking studies. Based on the inhibition constants of hHsp90 and PfHsp90 from the molecular docking studies, compounds TU-015 and TU-017 to TU-019 showed acceptable minimum energies in which all cases these energies were associated with low binding affinity (Ki) values (Table 3). More importantly, analysis of conformations and positions of TU-018 showed that these compounds exhibit a slight variability in terms of conformations inside the hHsp90 and PfHsp90 binding pockets (Figure 4). Previously, Wang and co-workers identified antimalarial compounds target a unique PfHsp90 hydrophobic pocket that extend from the lower edge of the conserved Hsp90 ATP binding pocket.41 It is possible that the compounds showing low binding affinity values in this study could be interacting with specific PfHsp90 hydrophobic pocket.42
CONCLUSIONS
In this work, we synthesized a focused series of resorcinol derivatives TU-014 to TU-019, initially developed as human Hsp90 inhibitors, and evaluated them for their activity against protozoan parasites P. falciparum and T.b. brucei, respectively. Although most compounds showed modest to weak activities, TU-018 displayed superior antiplasmodial and antitrypanosomal activity against both P falciparum and T.b. brucei (IC50 < 3 μM against both) without cytotoxicity against the HeLa cell line. Amongst other potential drug targets, the docking studies suggested that the antiprotozoal activity of these compounds could result from inhibition of Hsp90, an important molecular chaperone for survival of P falciparum and T.b. brucei protozoan parasites. Despite the limited small compound set, the preliminary structure-activity relationship (SAR) suggests that the isoxazole amide moiety consisting of non-aromatic heterocyclic ring systems provide extra interactions with Hsp90 enzyme and is critical for activity. In silico SwissADME prediction of the series indicated that these compounds possess acceptable ADMET and physicochemical properties. We believe that prosecuting these hits further against P. falciparum and T.b. brucei will increase the pool of quality starting points and spur further synthetic efforts in the quest for new antiprotozoal and antitrypanosomal agents. Finding new agents that act via new modes of action will have positive implications on the present treatment regiments that are under threat from emerging drug resistance, or at best, produce suboptimal therapeutic outcomes. Further optimization of these compounds through appropriate structural modifications core structure is currently underway in our labs and will be reported elsewhere.
ACKNOWLEDGMENTS
The authors acknowledge the financial support provided by Rhodes University Prestigious Scholarship (TU), Rhodes University Sandisa Imbewu (SDK and HCH), Rhodes University Research Committee (EDG) and the National Research Foundation of South Africa (SDK). The bioassay component of the project was funded by the South African Medical Research Council (MRC) and with funds from National Treasury under its Economic Competitiveness and Support Package.
SUPPLEMENTARY MATERIAL
Supplementary information for this article is provided as an online supplement. It contains experimental procedure, spectral characterisation data for the title compounds, including copies ofthe IR, MS, 1H NMR and 13C NMR spectra (Appendix B). The supplementary information file also included pLDH malaria (Figure S1) and cell cytotoxicity (Figure S2) assays.
SUPPLEMENTARY MATERIAL
Supplementary information for this article is provided in the online supplement.
ORCID IDs
Theoneste Umumararungu - https://orcid.org/0000-0002-5684-9607
Michelle Isaacs - https://orcid.org/0000-0002-0694-7487
Heinrich C Hoppe - https://orcid.org/0000-0003-4409-1955
Eleonora D Goosen - https://orcid.org/0000-0002-1470-9956
Setshaba D Khanye - https://orcid.org/0000-0003-0725-5738
REFERENCES
1. World malaria report 2021. Geneva: World Health Organization, 2021. Available online: https://www.who.int/publications/i/item/9789240040496 (accessed on 08 July 2022). [ Links ]
2. Petersen I, Eastman R, Lanzer M. Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 2011 Jun 6;585(11):1551-1562. https://doi.org/10.1016/j.febslet.2011.04.042. [ Links ]
3. Burrows JN, Burlot E, Campo B, Cherbuin S, Jeanneret S, Leroy D, Spangenberg T, Waterson D, Wells TNC, Willis P. Antimalarial drug discovery - the path towards eradication. Parasitology. 2014 Jan;141(1):128-139. https://doi.org/10.1017/S0031182013000826. [ Links ]
4. Wells TNC, Hooft van Huijsduijnen R, Van Voorhis WC. Malaria medicines: a glass half full? Nat Rev Drug Discov. 2015 Jun;14(6):424-442. https://doi.org/10.1038/nrd4573. [ Links ]
5. Kozlov M. Resistance to front-line malaria drugs confirmed in Africa. Nature. 2021;597(7878):604. https://doi.org/10.1038/d41586-021-02592-6. [ Links ]
6. Farooq F, Bergmann-Leitner ES. Immune escape mechanisms are Plasmodium's secret weapons foiling the success of potent and persistently efficacious malaria vaccines. Clin Immunol. 2015 Dec;161(2):136-143. https://doi.org/10.1016/jxlim.2015.08.015. [ Links ]
7. Medicines for Malaria Venture. Annual report 2020, 2021. https://www.mmv-annualreport2020.org/ (accessed on 13/07/22). [ Links ]
8. Andrews KT, Fisher G, Skinner-Adams TS. Drug repurposing and human parasitic protozoan diseases. Int J Parasitol Drugs Drug Resist. 2014 Mar 24;4(2):95-111. https://doi.org/10.1016/j.ijpddr.2014.02.002. [ Links ]
9. Gilbert IH. Target-based drug discovery for human African trypanosomiasis: selection of molecular target and chemical matter. Parasitology. 2014 Jan;141(1):28-36. https://doi.org/10.1017/S0031182013001017. [ Links ]
10. Rodgers J. Trypanosomiasis and the brain. Parasitology. 2010 Dec;137(14):1995-2006. https://doi.org/10.1017/S0031182009991806. [ Links ]
11. Franco JR, Simarro PP, Diarra A, Jannin JG. Epidemiology of human African trypanosomiasis. Clin Epidemiol. 2014 Aug 6;6:257-275. [ Links ]
12. Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov. 2006 Nov;5(11):941-955. https://doi.org/10.1038/nrd2144. [ Links ].
13. Tarral A, Blesson S, Mordt OV, Torreele E, Sassella D, Bray MA, Hovsepian L, Evène E, Gualano V, Felices M, et al. Determination of an optimal dosing regimen for fexinidazole, a novel oral drug for the treatment of human African trypanosomiasis: first-in-human studies. Clin Pharmacokinet. 2014 Jun;53(6):565-580. https://doi.org/10.1007/s40262-014-0136-3. [ Links ]
14. De Rycker M, Wyllie S, Horn D, Read KD, Gilbert IH. Anti-trypanosomatid drug discovery progress and challenges. Nat Rev Microbiol. 2023 21:31-50. https://doi.org/10.1038/s41579-022-00777-y [ Links ]
15. Eperon G, Balasegaram M, Potet J, Mowbray C, Valverde O, Chappuis F. Treatment options for second-stage gambiense human African trypanosomiasis. Expert Rev Anti Infect Ther. 2014 Nov;12(11):1407-1417. https://doi.org/10.1586/14787210.2014.959496. [ Links ]
16. Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, Jenks MX, Noe RA, Bowling TS, Mercer LT, et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl Trop Dis. 2011 Jun;5(6):e1151. https://doi.org/10.1371/journal.pntd.0001151. [ Links ]
17. Gelb MH, Van Voorhis WC, Buckner FS, Yokoyama K, Eastman R, Carpenter EP, Panethymitaki C, Brown KA, Smith DF. Protein farnesyl and N-myristoyl transferases: piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics. Mol Biochem Parasitol. 2003 Feb;126(2):155-163. https://doi.org/10.1016/S0166-6851(02)00282-7. [ Links ]
18. Pollastri MP, Campbell RK. Target repurposing for neglected diseases. Future Med Chem. 2011 Aug;3(10):1307-1315. https://doi.org/10.4155/fmc.11.92. [ Links ]
19. Njoroge M, Njuguna NM, Mutai P, Ongarora DSB, Smith PW, Chibale K. Recent approaches to chemical discovery and development against malaria and the neglected tropical diseases human African trypanosomiasis and schistosomiasis. Chem Rev. 2014 Nov 26;114(22):11138-11163. https://doi.org/10.1021/cr500098f. [ Links ]
20. Amata E, Xi H, Colmenarejo G, Gonzalez-Diaz R, Cordon-Obras C, Berlanga M, Manzano P, Erath J, Roncal NE, Lee PJ, et al. Identification of preferred human kinase inhibitors for sleeping sickness lead discovery. Are some kinases better than others for inhibitor repurposing? ACS Infect Dis. 2016 Mar 11;2(3):180-186. https://doi.org/10.1021/acsinfecdis.5b00136. [ Links ]
21. Kumar R, Musiyenko A, Barik S. The heat shock protein 90 of Plasmodium falciparum and antimalarial activity of its inhibitor, geldanamycin. Malar J. 2003 Sep 15;2(1):30. https://doi.org/10.1186/1475-2875-2-30.PMID:14514358. [ Links ]
22. Pallavi R, Roy N, Nageshan RK, Talukdar P, Pavithra SR, Reddy R, Venketesh S, Kumar R, Gupta AK, Singh RK, et al. Heat shock protein 90 as a drug target against protozoan infections: biochemical characterization of HSP90 from Plasmodium falciparum and Trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J Biol Chem. 2010 Dec 3;285(49):37964-37975. https://doi.org/10.1074/jbc.M110.155317. [ Links ]
23. Meyer KJ, Shapiro TA. Potent antitrypanosomal activities of heat shock protein 90 inhibitors in vitro and in vivo. J Infect Dis. 2013 Aug 1;208(3):489-499. https://doi.org/10.1093/infdis/jit179. [ Links ]
24. Debnath A, Shahinas D, Bryant C, Hirata K, Miyamoto Y, Hwang G, Gut J, Renslo AR, Pillai DR, Eckmann L, et al. Hsp90 inhibitors as new leads to target parasitic diarrheal diseases. Antimicrob Agents Chemother. 2014 Jul;58(7):4138-4144. https://doi.org/10.1128/AAC.02576-14. [ Links ]
25. Roy N, Nageshan RK, Ranade S, Tatu U. Heat shock protein 90 from neglected protozoan parasites. Biochim Biophys Acta. 2012 Mar;1823(3):707-711. https://doi.org/10.1016/j.bbamcr.2011.12.003. [ Links ]
26. Tatu U, Pallavi R, Yadav SC, Singh RK. Polynucleotide sequence, processes, composition and methods thereof [Patent]. WO/2011/154895A2. 2011. [ Links ]
27. Giannini G, Battistuzzi G. Exploring in vitro and in vivo Hsp90 inhibitors activity against human protozoan parasites. Bioorg Med Chem Lett. 2015 Feb 1;25(3):462-465. https://doi.org/10.1016/j.bmcl.2014.12.048. [ Links ]
28. Brasca MG, Mantegani S, Amboldi N, Bindi S, Caronni D, Casale E, Ceccarelli W, Colombo N, De Ponti A, Donati D, et al. Discovery of NMS-E973 as novel, selective and potent inhibitor of heat shock protein 90 (Hsp90). Bioorg Med Chem. 2013 Nov 15;21(22):7047-7063. https://doi.org/10.1016/j.bmc.2013.09.018. [ Links ]
29. Gumbo M, Beteck RM, Mandizvo T, Seldon R, Warner DF, Hoppe HC, Isaacs M, Laming D, Tam CC, Cheng LW, et al. Cinnamoyl-oxaborole amides: synthesis and their in vitro biological activity. Molecules. 2018 Aug 15;23(8):2038. https://doi.org/10.3390/molecules23082038. [ Links ]
30. Mbaba M, Mabhula AN, Boel N, Edkins AL, Isaacs M, Hoppe HC, Khanye SD. Ferrocenyl and organic novobiocin derivatives: synthesis and their in vitro biological activity. J Inorg Biochem. 2017 Jul;172:88-93. https://doi.org/10.1016/j.jinorgbio.2017.04.014. [ Links ]
31. Oderinlo OO, Tukulula M, Isaacs M, Hoppe HC, Taylor D, Smith VJ, Khanye SD. New thiazolidine-2,4-dione derivatives combined with organometallic ferrocene: Synthesis, structure and antiparasitic activity. Appl Organomet Chem. 2018;32(7):e4385. https://doi.org/10.1002/aoc.4385. [ Links ]
32. Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989 Dec;75(6):985-989. https://doi.org/10.2307/3282883. [ Links ]
33. Präbst K, Engelhardt H, Ringgeler S, Hübner H. Chapter 1, Basic colorimetric proliferation assays: MTT, WST, and resazurin. In: Gilbert D, Friedrich O, editors. Cell Viability Assays. Methods Mol. Biol. Vol. 1601. New York: Human Press; 2017. p. 1-17. https://doi.org/10.1007/978-1-4939-6960-9_1. [ Links ]
34. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010 Jan 30;31(2):455-461. [ Links ]
35. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017 Mar 3;7(1):42717. https://doi.org/10.1038/srep42717. [ Links ]
36. Makler MT, Ries JM, Williams JA, Bancroft JE, Piper RC, Gibbins BL, Hinrichs DJ. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am J Trop Med Hyg. 1993 Jun;48(6):739-741. https://doi.org/10.4269/ajtmh.1993.48.739. [ Links ]
37. Kar S, Leszczynski J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin Drug Discov. 2020 Dec;15(12):1473-1487. https://doi.org/10.1080/17460441.2020.1798926. [ Links ]
38. Bakchi B, Krishna AD, Sreecharan E, Ganesh VBJ, Niharika M, Maharshi S, Puttagunta SB, Sigalapalli DK, Bhandare RR, Shaik AB. An overview on application of SwissADME web tool in the design and development of anticancer, antitubercular and antimicrobial agents: A medicinal chemist's perspective. J Mol Struct. 2022;1259:132712. https://doi.org/10.1016/j.molstruc.2022.132712. [ Links ]
39. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001 Mar 1;46(1-3):3-26. https://doi.org/10.1016/S0169-409X(00)00129-0. [ Links ]
40. Corbett KD, Berger JM. Structure of the ATP-binding domain of Plasmodium falciparum Hsp90. Proteins. 2010 Oct;78(13):2738-2744. https://doi.org/10.1002/prot.22799. [ Links ]
41. Wang T, Bisson WH, Mäser P, Scapozza L, Picard D. Differences in conformational dynamics between Plasmodium falciparum and human Hsp90 orthologues enable the structure-based discovery of pathogen-selective inhibitors. J Med Chem. 2014 Mar 27;57(6):2524-2535. https://doi.org/10.1021/jm401801t. [ Links ]
42. Stofberg ML, Caillet C, de Villiers M, Zininga T. Inhibitors of the Plasmodium falciparum Hsp90 selective antimalarial drug design: the past, present and future. Cells. 2021 Oct 22;10(11):2849. https://doi.org/10.3390/cells10112849. [ Links ]
Received 29 August 2022
Revised 26 Febrauary 2023
Accepted 06 March 2023
* To whom correspondence should be addressed Email: s.khanye@ru.ac.za, umumararungut@yahoo.fr

