




![C-H activation: a Critical Evaluation of a Published Method and its Application Towards Inherently Chiral Calix[4]arenes](/img/en/next.gif)





Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.77 Durban 2023
http://dx.doi.org/10.17159/0379-4350/2023/v77a02
RESEARCH ARTICLE
Quinoline-benzofuran and quinoline-benzothiophene derivatives as antiplasmodium agents
Jonathan B HayI; Katherine A de VilliersI; Dale TaylorII; Tania OlivierI; Willem AL van OtterloI; Margaret AL BlackieI
IDepartment of Chemistry and Polymer Science, Stellenbosch University, Stellenbosch, South Africa
IIDivision of Clinical Pharmacology, Department of Medicine Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa
ABSTRACT
Two series of coupled heterocyclic systems have been synthesised and found to be efficacious against the NF54 chloroquine sensitive strain of P. falciparum in an in vitro assay. Quinolines were coupled with benzofurans in the first series and benzothiophenes in the second series. Compounds with an amide linkage are more efficacious by one order of magnitude than the ester equivalent. No other clear pattern was discernible. Compounds were also tested for toxicity using an MTT assay. All compounds tested showed selectivity towards P. falciparum. Barring one, all compounds tested showed greater efficacy than chloroquine against β-hematin inhibition. But no correlation was observed between β-hematin inhibition and efficacy against P. falciparum.
Keywords: 4-aminoquinoline, β-hematin, benzofuran, benzothiophene, Plasmodium falciparum
INTRODUCTION
The search for new chemical entities as potential antiplasmodium drugs continues unabated due to parasite resistance to many clinical antimalarial drugs. There are various approaches which can be taken to this end. In this study we have chosen to combine two biologically privileged scaffolds - a quinoline and either a benzofuran or a benzothiophene. We then used a phenotypic approach to ascertain efficacy against the asexual blood stage of Plasmodium falciparum. Furthermore, to probe activity against the heme detoxification pathway of P. falciparum, we have determined the efficacy against synthetic hemozoin (β-hematin) formation.
In an earlier study, we had explored the synthesis of indoles, benzimidazoles, benzoxazoles and benzothiophenes coupled to either a phenyl or naphthalenyl group (the most active of the benzothiophene derivatives, compound 1, is shown in Figure 1).1 With the exception of the benzoxazoles, all compounds showed efficacy against the NF54 strain of P. falciparum in the order of 100 nM to 10 μM. It was notable that the benzoxazoles proved to be completely inactive against the NF54 strain of P. falciparum. The motivation for this work was to determine whether benzofuran and benzothiophene derivatives have similar efficacy. The motivation of switching from the phenyl or naphthalenyl group to the quinolinyl derivatives was to increase the likelihood of at least some efficacy towards P. falciparum. The reason for this choice was because of the known efficacy of the quinolinyl moiety against P. falciparum.2
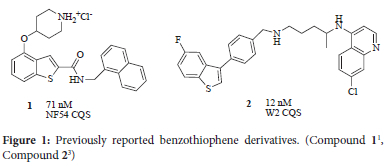
In previous work by Konstantinovic et al., benzothiophenes have been coupled with 7-chloroquinolines. For example compound 2 shown in Figure 1 gave an IC50 value of 12 nM against W2, a chloroquine resistant strain of P. falciparum.3 Furthermore, benzofuran has been established as a biologically active molecule4 and has been incorporated into active antiplasmodials, 5 although to our knowledge it has not been coupled with 7-chloroquinoline.
The substitution pattern on the benzothiophene was retained from our previous work1 That is to say the coupling linkage through either the amide or ester at position 2 and the ether linkage at position 4 as illustrated by compound 1. In the new series of compounds reported herein various benzyl derivatives were used instead of the piperidine moiety. In addition, the naphthalenyl group was substituted with both a 4-amino-7-chloroquinoline and a 4-amino-7-trifluoromethyl-quinoline. Electron-withdrawing groups in the 7-position of the quinoline have long been associated with increased efficacy for compounds that utilise haemozoin inhibition as a mode of action.6 The two fused heterocyclic moieties were linked using amide and ester bonds in turn, giving the general structure as shown in Figure 2.
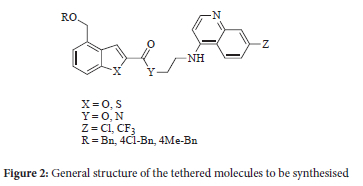
In this series of compounds there are several points of difference in the molecules, which may or may not influence efficacy against P. falciparum.
1. Benzothiophene and benzofuran - in our previous work1 there was a significant loss of efficacy with the benzoxazole moiety.
2. Amide and ester linkage - in our previous work1 there was no discernible pattern on the basis of the linkage alone. This is probably not surprising as the linkage is not likely to interfere significantly with the mode of action of the compounds. Although it is known that ester linkages are more prone to failure under biological conditions, this may not have an impact in an in vitro study.
3. Substitution on the para position of the benzyl moiety - this variation is not building on prior work and so the impact of variation in this position was unknown at the time of synthesising the molecules.
4. Chloro and trifluoromethyl groups on the quinoline - both groups are electron-withdrawing and have been shown to be efficacious against synthetic haemozoin (p-haematin) inhibition and P. falciparum.6
This series of compounds is therefore based on a phenotypic approach rather than a target based approach. That is to say we have combined moieties which have shown efficacy against P. falciparum.
RESULTS AND DISCUSSION
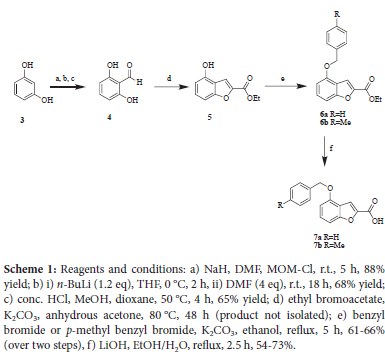
The synthesis of the benzofuran scaffold 5 was achieved starting from resorcinol 3 (Scheme 1). The installation of the aldehyde 4 was achieved via ort/îo-formylation, which required protection and deprotection of the two hydroxyl groups using a MOM ether group.7 Reaction of 4 with ethyl bromoacetate via a modified Rap-Stoemer reaction yielded benzofuran 5 with the appropriate handles for further substitution in positions 2 and 4.7 Attempted isolation of product 5 consistently resulted in a mixture of an intermediate and the desired product. The basic conditions of the installation of the benzyl group proved to be sufficient to effect full cyclisation. Sufficient amounts of the pure product were isolated for characterisation, but as a matter of course compound 5 was not usually isolated. The acetone was removed under reduced pressure and the crude mixture carried through to the next step. The yield over the two steps of 66% was deemed acceptable. The appearance of a singlet at 7.67 ppm integrating for one proton assigned to position 3 of the benzofuran in the proton NMR spectrum confirmed the successful synthesis of the heterocycle. Benzylation was then carried out to attach an aromatic ring. Benzyl bromide and p-methylbenzyl bromide were used to afford 6a and 6b in reasonable yield (61-66%). The ethyl group was then removed in anticipation of the amidation or esterification reactions used to couple the benzofuran to a quinoline moiety (see later).8
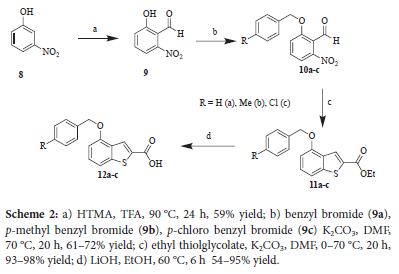
Synthesis of the benzothiophene was achieved using a slightly different approach, since the benzyl group could serve as the protecting group for the phenol prior to ortho-formylation.
In this case the starting material was 3-nitrophenol, 8, Scheme 2. The installation of the aldehyde ortho to the phenol was achieved in reasonable yield using an established procedure.9 However, regioselectivity was an issue, and the isomer with the aldehyde para to the nitro group was an unavoidable by-product using this method. However, the two isomers were separable via column chromatography and the method was deemed acceptable. The phenol was then benzylated using benzyl bromide (10a), p-methyl benzyl bromide (10b) or p-chloro benzyl bromide (10c). The final step was the formation of the benzothiophene using ethyl thioglycolate. Again the characteristic single proton signal at 8.27 ppm in the proton NMR spectra indicated that the benzothiophene had indeed formed. Further characterisation using 13C NMR and IR spectroscopy, as well as HRMS, confirmed that products 11a-c had been formed.
The final part of the synthesis was the coupling for the benzofurans (7a-b) or benzothiophenes (12a-c) to 4-amino-7-chloroquinoline or 4-amino-7-trifluoromethylquinoline (Scheme 3). The reactions began with the 4,7-dichloroquinoline 13a or 4-chloro-7-trifluoromethylquinoline 13b, on which the reaction with 1,2-diaminoethane was carried out under reflux. This was followed by a CDI-mediated coupling between the free acid derivatives of both benzofuran and benzothiophene moieties.10
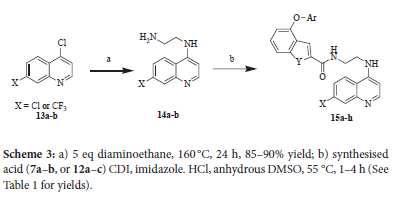
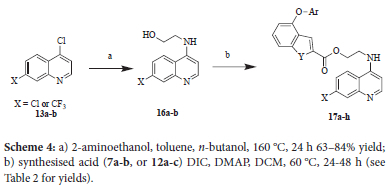
The quinolines, 13a and 13b, were also reacted with 2-aminoethanol at 160 °C in the presence of a mixture of toluene and n-butanol in a sealed microwave vial, which was effectively used as a pressure vessel (Scheme 4). The coupling to the acid derivatives of the benzofuran or benzothiophene moiety was then achieved via a modification of the Stiglich esterification.11 Instead of using DCC as the coupling reagent, DIC was used as it was readily available.
Characterisation of compounds 15a-h and 17a-h were confirmed using 1H and 13C NMR and IR spectroscopy, as well as HRMS.
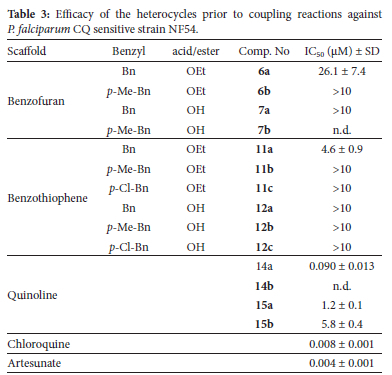
Biological results
Prior to coupling, the benzofurans, benzothiophenes and quinolines were tested for antiplasmodium efficacy. The activity of the quinolines are known6 and the values reported here fall within the expected range of activity for these types of compounds. For the most part, neither the benzofurans nor the benzothiophenes demonstrated antiplasmodium efficacy within the range of concentrations of the assay. Under the mildly acidic conditions of the food vacuole of P. falciparum, the free acids (7a-b and 12a-c) may react to form esters or amides with alcohol or amine groups in the system, so their poor efficacy was anticipated. An interesting result was the moderate activity observed for the unsubstituted benzyl-containing esters (6a and 11a). Although the activity is 500-3000 fold weaker than chloroquine this raises questions about the potential mode of action which, unfortunately, cannot be answered within the scope of the series of compounds that have been synthesised. Nonetheless, this result points to the value of conducting the activity assay on the individual heterocyclic fragments, and not just the final products.
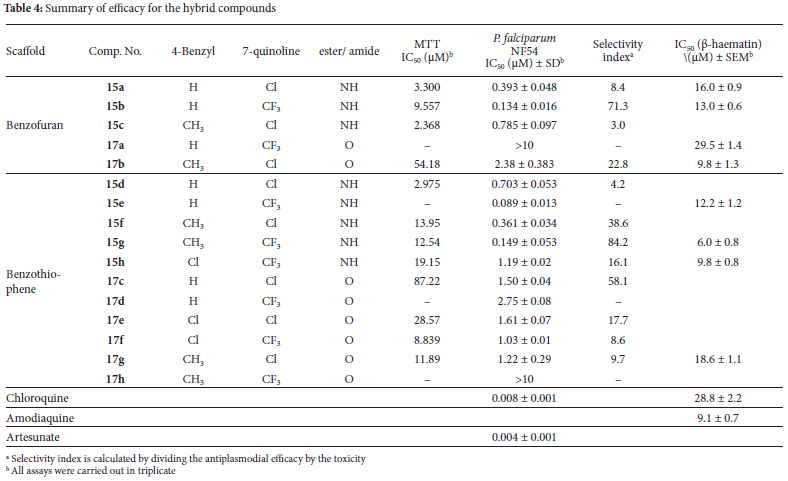
For hybrid compounds where we had sufficient material, testing was done against the NF54 chloroquine sensitive strain of P. falciparum, toxicity was determined and efficacy in a NP-40 detergent mediated p-haematin inhibition assay was evaluated. All assays were carried out in triplicate. The data are summarized in Table 4.
From the activity data in Table 4, there is no discernible pattern with respect to the benzofuran and benzothiophene moieties. That is to say that it is not evident that substituting the sulfur for the oxygen atom makes any substantial difference to the efficacy of these compounds. The increase in efficacy associated with the unsubstituted benzyl group which was observed in the fragment assay is not evident in the hybrid compounds. There is also no clear correlation between p-hematin inhibition activity and antiplasmodial efficacy. This lack of correlation is noteworthy because in smaller molecules, the 7-chloroquinoline scaffold tends to have a consistent greater efficacy than the 7-trifluoromethylquinoline core.6 This is not the case here; for example, in the compound pairs 15d/15e and 15f/15g, the 7-trifluoromethyl substituted compound has greater efficacy in both cases than the 7-chloro hybrid.
However, the most consistent trend comes from the variation in the ester and amide linkages. In all cases, the compound with the amide linkage is at least one order of magnitude more efficacious than the ester counterpart. It is not at all clear at this stage why this factor should make such a difference. It may be that the conditions of the assay are such that the ester linkage is in fact more labile than we anticipated. This question is worth further investigation.
EXPERIMENTAL
Chemical reagents used for synthesis were either obtained from the chemical store or purchased from Sigma-Aldrich and used without purification. Reaction solvents used for synthesis were all purchased from Sigma-Aldrich/Merck with purity grades > 98%. The procedures for drying reaction solvents were obtained as described in Armarego et al.12 Solvents used for chromatographic purposes (ethyl acetate, hexane, dichloromethane, methanol, ethanol and acetone) were obtained from the chemical store and distilled by conventional method at their respective boiling points to remove impurities.
Thin layer chromatography (TLC) was performed using Macherey-Nagel Alugram* Xtra SIL G/UV254 silica gel 60 coated aluminium plates. TLC visualization was carried out using either a UV lamp with two wavelength options (254 and 365 nm) or with liquid stains to develop the plate. To visualize the components of reaction mixtures, bromocresol green was used for acidic compounds, potassium permanganate (KMnO4) as general-purpose stain and p-Anisaldehyde #1 (PAA #1) as specialized stain for functional group change identification.1 Flash column chromatography was carried out using Merck silica gel (230-400 mesh) either by conventional method or automated instrument. A Teledyne Isco CombiFlash Rf150 automated purification instrument, fitted with a variable 200-400 nm UV detector, was used with reusable columns of varying sizes.
Nuclear magnetic resonance spectra were recorded using a 300 MHz Varian VNMRS (75 MHz for 13C), a 400 MHz Varian Unity Inova (101 MHz for 13C) and a 600 MHz Varian Unity Inova (151 MHz for 13C and 262 MHz for 19F). Mass spectroscopy (MS) and purity analysis was carried out on a Waters SYNAPT G2 mass spectrometer fitted with a Time-of-Flight (TOF) mass analyzer using the Electrospray Ionization positive or negative (ESI+ or ESI-) method. Infrared (IR) spectra were collected on a Thermo Fisher Scientific Nicolet iS10 FTIR photometer and setup to collect the spectra by double beam Fourier Transform Infrared Attenuated Total Reflection (FTIR-ATR) technique and peak values units reported in wavenumber (cm-1). Melting points were obtained using a Gallenkamp Melting Point Apparatus and temperatures measured to the first decimal in degrees Celsius (°C).
2,6-Dihydroxybenzaldehyde 4
Compound 4 was synthesized in three steps using the method described by Masubuchi et al. 7
The crude mixture was then purified by automated column chromatography (Rf = 0.61, 15% EtOAc/Hex) to afford the product as a yellow solid (542 mg, 3.93 mmol, 65%). (mp 156.4-159.1 °C); 1H NMR (400 MHz, CDCl3) Ô 10.72 (broad s, 2H), 10.17 (s, 1H), 7.09 (t, J 8.2 Hz, 1H), 6.19 (d, J 8.2 Hz, 2H); 13C NMR (101 MHz, CDCl3) Ô 194.4 (2C), 162.3, 138.3, 110.3 106.8 (2C); vmax 2885 (br w), 1617(s), 1514, 1460, 1232 (s), 1202, 1156, 710 (s). This is a known compound and values correspond well with previously reported values. 7
Ethyl 4-hydroxybenzofuran-2-carboxylate 5
Dry acetone (6 mL) and anhydrous potassium carbonate (368 mg, 2.66 mmol) were added to a 25 mL microwave vial, equipped with a stir bar, and stirred at 0 °C. 2,6-Dihydroxybenzaldehyde (122 mg, 0.888 mmol) was added as a single portion and stirred until completely dissolved. Then ethyl bromoacetate (0.11 mL, 0.16 g, 0.98 mmol) was diluted with dry acetone (0.5 mL) and added dropwise to the reaction vial. The vial was removed from the ice bath, sealed with a vial cap and septum, then heated to 100 °C for 48 hours. The reaction was monitored by TLC for completion during the reaction period and after 48 hours the vial was cooled to room temperature and an additional portion of anhydrous potassium carbonate (195 mg, 1.41 mmol) added. The vial was resealed and heated for a further 24 hours. After completion, the reaction mixture was cooled to room temperature and diluted with distilled water (10 mL), followed by acidification with 1 N HCl to a pH of ~5. The aqueous mixture was extracted with ethyl acetate (3 x 15 mL), the organic layers combined and washed with water (20 mL) and then brine (10 mL). The organic layer was collected, dried over anhydrous magnesium sulfate and the solution collected by vacuum filtration. The ethyl acetate was collected and evaporated under reduced pressure to afford the crude product as a yellow oil. The crude product was purified by automated column chromatography and the pure product obtained as a pale-yellow powder (55.8 mg, 0.248 mmol, 28%). Rf = 0.35, 15% EtOAc/Hex. (mp 136-138 °C); 1H NMR (400 MHz, CDCl3) Ô 7.72 (s, 1H), 7.32-7.23 (m, 1H), 7.14 (d, J 8.4 Hz, 1H), 6.70 (d, J 7.8 Hz, 1H), 6.18 (s, 1H), 4.45 (q, J 7.1 Hz, 2H), 1.42 (t, J 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.1, 157.3, 150.9, 144.4, 128.7, 117.0, 111.7, 108.5, 105.0, 61.8, 14.4; vmax 3297 (w), 1690, 1605, 1557, 1191 (s), 1109, 1046, 844, 754 (s), 727; HRMS-TOF ESI+: m/z [M+H]+ calculated for C11H11O4: 207.0658; found: 207.0661. This is a known compound and values correspond well with previously reported values. 7
SYNTHESIS OF 6A-B
General procedure
A minimal amount of dry acetone, anhydrous potassium carbonate (0.5 to 2.1 equivalents) and 2,6-dihydroxybenzaldehyde (1 equivalent) were added in a single portion in a round-bottom flask. The mixture was left to stir at room temperature until the benzaldehyde was completely dissolved. While stirring, ethyl bromoacetate (1 equivalent), diluted in an equal amount of dry acetone, was added dropwise to the reaction flask. The reaction mixture was refluxed for 48 hours and monitored by TLC for completion. On completion, the reaction mixture was cooled to room temperature and the solvent evaporated under reduced pressure. The concentrated reaction residue was dissolved in dry ethanol (10 mL) and anhydrous potassium carbonate (3 equivalents) added. 4-Methylbenzyl chloride (0.7 equivalent) or benzyl bromide (1 equivalent) was added to the stirring mixture at room temperature and then heated under reflux for 6 hours, monitoring completion by TLC. On completion, the reaction mixture was cooled to room temperature and the solvent evaporated under reduced pressure. The residue was dissolved in water and extracted with ethyl acetate (3 X 25 mL), the organic layers combined and washed once with water followed by brine. The organic layer was collected, dried over anhydrous magnesium sulfate and the solution collected by vacuum filtration. The solvent was evaporated under reduced pressure the crude product was obtained as yellow to brown oils.
Ethyl 4-(benzyloxy)benzofuran-2-carboxylate 6a
The general procedure described above was followed using dry acetone (15 mL), anhydrous potassium carbonate (90 mg, 0.65 mmol, 2,6-dihydroxybenzaldehyde (160 mg, 1.15 mmol), ethyl bromoacetate (0.128 mL, 193 mg, 1.15 mmol), benzyl bromide (o.138 mL, 198 mg, 1.15 mmol) and anhydrous potassium carbonate (480 mg, 3.47 mmol). The crude product was obtained as a yellow oil and purified as described in the general procedure to afford the product as a white solid (159 mg, 0.537 mmol, 61%). Rf = 0.50, 15% EtOAc/Hex; 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.52-7.31 (m, 6H), 7.24-7.18 (m, 1H), 6.78-6.72 (m, 1H), 5.21 (s, 2H), 4.43 (q, J 7.1 Hz, 2H), 1.42 (t, J 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) Ô 159.6, 157.0, 153.8, 144.6, 136.6, 128.7 (2C) 128.6, 128.2, 127.5 (2C), 118.3, 111.8, 105.5, 104.9, 70.4, 61.5, 14.4; vmax 2913 (w), 1757, 1732, 1715 (m), 1561, 1334, 1177, 1021, 782; HRMS-TOF ESI+: m/z [M+H] + calculated for C18H17O4: 297.1129; found: 297.1127.
Ethyl 4-[(4-methylbenzyl)oxy]benzofuran-2-carboxylate 6b
The general procedure described above was followed using dry acetone (15 mL), anhydrous potassium carbonate (674 mg, 4.88 mmol, 2,6-dihydroxybenzaldehyde (321 mg, 2.32 mmol), ethyl bromoacetate (0.26 mL, 390 mg, 2.3 mmol), 4-methylbenzyl chloride (0.22 mL, 0.23 g, 1.6 mmol) and anhydrous potassium carbonate (670 mg, 4.84 mmol). The crude product was obtained as a yellow oil and purified as described in the general procedure to afford the product as a white solid (341 mg, 1.09 mmol, 66%). Rf = 0.50, 15% EtOAc/Hex. (mp 78-80 °C)1H NMR (400 MHz, CDCl3) Ô 7.67 (s, 1H), 7.38-7.31 (m, 3H), 7.24-7.17 (m, 3H), 6.75 (d, J 8.0 Hz, 1H), 5.16 (s, 2H), 4.43 (q, J 7.1 Hz, 2H), 2.38 (s, 3H), 1.41 (t, J 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 159.7, 157.0, 153.9, 144.5, 138.1, 133.6, 129.4 (2C), 128.6, 127.7 (2C), 118.3, 111.9, 105.7, 104.9, 70.4, 61.5, 21.3, 14.4; vmax 2916, 2848, 1732, 1460, 1331, 1257, 1169, 1120, 1109, 1082, 1018, 807, 780, 759, 731, 718; HRMS-TOF ESI+: m/z [M+H]+ calculated for C19H19O4: 311.1284; found: 311.1283.
Synthesis of 7a-b
General Procedure
A minimal amount of ethanol, distilled water, 6a or 6b (1 equivalent) and lithium hydroxide (3 to 4 equivalents) were added in a round-bottom flask and stirred. The mixture was left to stir under reflux for 2.5 hours, monitoring completion by TLC using bromocresol green as the visualizing stain. On completion, the reaction mixture was cooled to room temperature and the solvent evaporated under reduced pressure. The residue was diluted with water and acidified with 2N HCl solution until a white precipitate formed. The precipitate was collected by filtration, the solids dissolved using ethyl acetate and dried over anhydrous magnesium sulfate. The magnesium sulfate was removed by vacuum filtration and the ethyl acetate evaporated under reduced pressure to afford the products as white solids (54-72%).
4-(Benzyloxy)benzofuran-2-carboxylic acid 7a
The general procedure described above was followed using ethanol (15 mL), distilled water (1 mL), 6a (1.00 g, 3.37 mmol) and lithium hydroxide (242 mg, 10.1 mmol). The product was purified as described above and afforded the product as a white solid (489 mg, 1.82 mmol, 54%). (mp 124-126 °C); 1H NMR (400 MHz, CDCl3) δ 7.60 (s, 1H), 7.46-7.39 (m, 2H), 7.38-7.25 (m, 4H), 7.17-7.10 (m, 1H), 6.73-6.66 (m, 1H), 5.16 (s, 2H); 13C NMR (101 MHz, CDCl3) Ô 161.0, 156.7, 153.5, 144.9, 136.3, 128.4, (2C), 128.1, 127.9, 127.1 (2C), 118.1, 111.2, 105.1, 104.7, 70.1; vmax 2848 (v br), 1682, 1567, 1255 (s), 1197, 916 (m), 779, 751 (m), 695; HRMS-TOF ESI+: m/z [M+H]+ calculated for C16H13O4: 269.0819; found: 269.0814.
4-[(4-Methylbenzyl)oxy]benzofuran-2-carboxylic acid 7b
The general procedure described above was followed using ethanol (10 mL), distilled water (1 mL), 6b (200 mg, 0.643 mmol) and lithium hydroxide (56.3 mg, 2.35 mmol). The product was purified as described above and afforded the product as a white solid (125 mg, 0.443 mmol, 72%). 1H NMR (400 MHz, (CD3)2SO) δ 7.50 (s, 1H), 7.31-7.22 (m, 3H), 7.14-7.04 (m, 3H), 6.69 (d, J 8.0 Hz, 1H), 5.09 (s, 2H), 2.28 (s, 3H); 13C NMR (101 MHz, (CD3)2SO) δ 160.4, 156.1, 153.1, 144.7, 137.2, 132.9, 128.7 (2C), 127.8, 127.0 (2C), 117.6, 110.6, 104.5, 69.6, 39.5, 20.6; vmax 2916 (v br), 1679 (s), 1557 (m), 1499, 1194 (s), 1108, 1070, 923 (m), 780, 758 (s); HRMS-TOF ESI+: m/z [M+H] + calculated for C17H15O4: 283.0971; found: 283.0971.
2-Hydroxy-6-nitrobenzaldehyde 9
Hexamethylenetetramine (2.44 g, 17.3 mmol), 3-nitrophenol (2.01 g, 14.4 mmol) and trifluoroacetic acid (16.0 mL, 23.9 g, 209.5 mmol) were combined in a Schlenk tube. The reaction vessel was sealed under nitrogen atmosphere and stirred for 24 hours at 90 °C. The reaction mixture was then cooled to room temperature, distilled water (30 mL) added and the mixture stirred for a further 3 hours and completion monitored by TLC. On completion, the reaction mixture was diluted with water (150 mL) and extracted with ethyl acetate (5 x 30 mL). The organic layers were combined, washed with water (5 x 50 mL) and the organic layer washed with brine (50 mL). The organic layer was dried over anhydrous magnesium sulfate, the solvent collected by vacuum filtration and concentrated under reduced pressure. The crude product was obtained as a thick orange oil and purified by automated column chromatography to afford the product as a yellow solid (1.42 g, 8.49 mmol, 59%). Rf = 0.58, 15% EtOAc/Hex. (mp 52-54°C); 1H NMR (400 MHz, CDCl3) Ô 12.11 (s, 1H), 10.33 (s, 1H), 7.65-7.60 (m, 1H), 7.57-7.54 (m, 1H), 7.32-7.28 (m, 1H); vmax 3216 (w br), 1672 (s), 1532; This is a known compound and values correspond well with previously reported values.1
SYNTHESIS OF 10A-C
General procedure
Dry DMF was combined with anhydrous potassium carbonate (3-4 equivalents) in a round-bottom flask and 9 (1 equivalent) added. The solution was stirred at room temperature and the respective 4-substituted-benzyl halides (1.3-2 equivalents) added dropwise. The reaction mixture was then heated to 70 °C and stirred for 20 hours where on completion the mixture was cooled to room temperature. The reaction mixture was diluted with distilled water (100 mL) and acidified to a pH ~6 with 1N HCl. The aqueous mixture was extracted with ethyl acetate (4 x25 mL), the organic layers combined, washed with distilled water (3 x 40 mL) and then with brine (30 mL). The organic layer was collected, dried over anhydrous magnesium sulfate and the solvent collected by vacuum filtration. The solvent was evaporated under reduced pressure and the crude product purified by automated column chromatography (15% EtOAc/Hex). Residual DMF was removed by freeze-dryer to afford the pure products as white solids (61-72%).
2-(Benzyloxy)-6-nitrobenzaldehyde 10a
The general procedure described above was followed using dry DMF (20 mL), anhydrous potassium carbonate (2.30 g, 31.6 mmol), 9 (1.32 g, 7.90 mmol) and benzyl bromide (1.41 mL, 2.03 g, 11.8 mmol). The product was obtained as a white solid (1.44 g, 5.60 mmol, 71%). Rf = 0.30, 15% EtOAc/Hex. 1H NMR (400 MHz, CDCl3) δ 10.42 (s, 1H), 7.59-7.52 (m, 1H), 7.39 (m, 6H), 7.31-7.26 (m, 1H), 5.21 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 187.7, 158.9, 135.1, 133.6, 128.9, 127.3, (2C), 121.0, 117.7, 115.8, 71.6. vmax 2876 (w br), 1690 (s), 1520, 1396, 1347, 1247 (s), 1178, 1002, 871, 811, 737, 688. This is a known compound and values correspond well with previously reported values. 1
2-[(4-Methylbenzyl)oxy]-6-nitrobenzaldehyde 10b
The general procedure described above was followed using dry DMF (30 mL), anhydrous potassium carbonate (2.55 g, 18.5 mmol), 9 (1.03 mg, 6.16 mmol) and 4-methylbenzyl chloride (1.06 mL, 1.12 g, 8.01 mmol). The product was obtained as a white solid (1.20 g, 4.42 mmol, 72%). Rf = 0.31, 15% EtOAc/Hex. 1H NMR (400 MHz, CDCl3) δ 10.41 (s, 1H), 7.58-7.51 (m, 1H), 7.43-7.38 (m, 1H), 7.31-7.24 (m), 7.237.18 (m, 2H), 5.17 (s, 2H), 2.36 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 187.7, 159.1, 148.8, 138.6, 133.6, 132.1, 129.6 (2C), 127.5 (2C), 121.0, 117.7, 115.8, 71.6, 21.3; vmax 2917, 2862 (w br), 1697 (s), 1519, 1499, 1469, 1345, 1275 (s), 1177, 1055, 796, 737.
2-[(4-Chlorobenzyl)oxy]-6-nitrobenzaldehyde 10c
The general procedure described above was followed using dry DMF (30 mL), anhydrous potassium carbonate (2.42 g, 17.5 mmol), 9 (977 mg, 5.84 mmol) and 4-chlorobenzyl chloride (0.96 mL, 1.2 g, 7.6 mmol). The product was obtained as a white solid (1.03 mg, 3.54 mmol, 61%). Rf = 0.18, 15% EtOAc/Hex. 1H NMR (400 MHz, CDCl3) Ô 10.40 (s, 1H), 7.56 (m, 1H), 7.50-7.45 (m, 1H), 7.39-7.31 (m, 4H), 7.29-7.23 (m, 1H), 5.17 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 187.6, 158.4, 134.6, 133.6, 133.5, 129.2 (2C), 128.7 (2C), 121.5, 117.7, 116.2, 70.9. vmax 2916, 2864 (w br), 1705 (s), 1522, 1343, 1283, 1054, 812, 737.
Synthesis of 11a-c
General procedure
Dry DMF, anhydrous potassium carbonate (2.5 equivalents) and 10a, 10b or 10c (1 equivalent) were combined in a round-bottom flask. The solution was stirred at 0 °C, ethyl thioglycolate (1.25-1.5 equivalents) added dropwise and the reaction mixture stirred for 30 minutes. The reaction mixture was then heated to 70 °C and stirred for 24 hours where on completion the mixture was cooled to room temperature. The reaction mixture was diluted with distilled water and acidified to a pH ~6 with 1N HCl. The aqueous mixture was extracted with ethyl acetate (4 x 25 mL), the organic layers combined, washed with distilled water (3 x 40 mL) and then with brine (30 mL). The organic layer was collected, dried over anhydrous magnesium sulfate and the solvent collected by vacuum filtration. The solvent was evaporated under reduced pressure and the crude product purified by automated column chromatography (15% EtOAc/Hex). Residual DMF was removed by freeze-dryer to afford the pure products as white solids (93-98%).
Ethyl 4-(benzyloxy)benzo[b]thiophene-2-carboxylate 11a
The general procedure was followed using 10a (167 mg, 0.649 mmol), dry DMF (5 mL), anhydrous potassium carbonate (224 mg, 1.62 mmol) and ethyl thioglycolate (0.10 mL, 11 mg, 0.92 mmol). The pure product was obtained as a white solid (199 mg, 0.638 mmol, 98%). Rf = 0.73, 15% EtOAc/Hex. 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 7.52-7.47 (m, 2H), 7.46-7.32 (m, 5H), 6.81 (d, J 7.2 Hz, 1H), 5.21 (s, 2H), 4.41 (q, J 7.1 Hz, 2H), 1.42 (t, J 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 189.3, 162.8, 155.3, 143.8, 136.5, 132.2, 130.2, 128.7 (2C), 128.2, 128.1, 127.4 (2C), 115.1, 105.4, 70.1, 61.5, 14.4; vmax 2984, 1697 (s), 1562, 1518, 1463, 1284 (s), 1254 (m), 1149, 1071, 1016, 749; HRMS-TOF ESI+: m/z [M+H] + calculated for C18H17O3S: 313.0899; found: 313.0898. This is a known compound and values correspond well with previously reported values.1
Ethyl 4-[(4-methylbenzyl)oxy]benzo[b]thiophene-2-carboxylate 11b
The general procedure was followed using 10b (154 mg, 0.568 mmol), DMF (10 mL), anhydrous potassium carbonate (207 mg, 1.44 mmol) and ethyl thioglycolate (0.10 mL, 11 mg, 0.92 mmol). The pure product was obtained as a white solid (172 mg, 0.527 mmol, 93%). Rf = 0.71, 15% EtOAc/Hex. (mp 104-106°C); 1H NMR (400 MHz, CDCl3) Ô 8.27 (s, 1H), 7.45-7.36 (m, 4H), 7.26-7.21 (m, 2H), 6.82 (d, J 7.7 Hz, 1H), 5.17 (s, 2H), 4.40 (q, J 7.1 Hz, 2H), 2.39 (s, 3H), 1.41 (t, J 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) Ô 162.9, 155.5, 143.9, 138.0, 133.6, 132.2, 130.3, 129.4 (2C), 128.3, 127.7 (2C), 127.6, 115.1, 105.5, 70.2, 61.5, 21.3, 14.4; vmax 2917, 1695 (s), 1561, 1516, 1462, 1303 (s), 1285, 1253 (m), 1150, 749; HRMS-TOF ESI+: m/z [M+H]+ calculated for C19H19O3S: 327.1059; found: 327.1055.
Ethyl 4-[(4-chlorobenzyl)oxy]benzo[b]thiophene-2-carboxylate 11c
The general procedure was followed using 10c (200 mg, 0.686 mmol), DMF (10 mL), anhydrous potassium carbonate (237 mg, 1.71 mmol) and ethyl thioglycolate (0.08 mL, 90 mg, 0.7 mmol). The pure product was obtained as a white solid (226 mg, 0.653 mmol, 95%). Rf = 0.61, 15% EtOAc/Hex. 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.497.32 (m, 6H), 6.77 (d, J 7.8 Hz, 1H), 5.17 (s, 2H), 4.40 (q, J 7.1 Hz, 2H), 1.41 (t, J 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) Ô 162.9, 155.1, 143.9, 135.1, 134.0, 132.4, 130.2, 128.9 (2C), 128.8 (2C), 128.2, 127.3, 115.4, 105.5, 69.4, 61.6, 14.4; vmax 2917, 1688 (s), 1563, 1285 (s), 1253 (m), 1149, 1073, 1022, 810, 746; HRMS-TOF MS ESI+: m/z [M+H] + calculated for C18H16ClO3S: 347.0509; found: 347.0518.
Synthesis of 12a-c
General Procedure
Ethanol, lithium hydroxide (5-10 equivalents) and one ofthe respective derivatives 11a-c (1 equivalent) were added to a round-bottom flask. The reaction mixture was heated to 60 °C and stirred for 6 hours. On completion the ethanol was removed under reduced pressure, the residue diluted with water and basified with 1N NaOH. The aqueous mixture was extracted with ethyl acetate (3 x 15 mL) and the organic layers kept aside. The aqueous layer was then acidified with 1N HCl and extracted with ethyl acetate (3 x 15 mL). The organic layers were combined, dried over anhydrous magnesium sulfate and the solvent collected by vacuum filtration. The ethyl acetate was removed under reduced pressure to afford the pure products as white solids.
4-(Benzyloxy)benzo[b]thiophene-2-carboxylic acid 12a
The general procedure was followed using 11a (200 mg, 0.640 mmol), ethanol (15 mL) and lithium hydroxide (83.0 mg, 3.46 mmol). The pure product was obtained as a white solid (97.8 mg, 0.344 mmol, 54%). Rf = 0.11, 15% EtOAc/Hex. (mp 178-180 °C); 1H NMR (600 MHz, CDCl3) Ô 10.13 (broad s, 1H), 8.17 (s, 1H), 7.40-7.35 (m, 2H), 7.32-7.20 (m, 5H), 6.69 (d, J 7.8 Hz, 1H), 5.07 (s, 2H); 13C NMR (151 MHz, CDCl3) δ 165.5, 154.9, 143.6, 136.4, 134.1, 130.1, 128.3 (2C), 127.8, 127.6, 127.1 (2C), 126.8, 114.8, 105.1, 69.7; vmax 2483, 1667 (s), 1562, 1518, 1257 (s), 1153 (s), 1060;HRMS-TOF MS ESI+: m/z [M+H] + calculated for C16H13O4 285.0586; found: 285.0579.
4-[(4-Methylbenzyl)oxy]benzo[b]thiophene-2-carboxylic acid 12b
The general procedure described above was followed using 11b (501 mg, 1.54 mmol), ethanol (30 mL) and lithium hydroxide (362 mg, 15.2 mmol). The pure product was obtained as a white solid (438 mg, 1.47 mmol, 96%). Rf = 0.10, 15% EtOAc/Hex. (mp 127-129 °C); 1H NMR (400 MHz, (CD3)2 SO) δ 8.11 (s, 1H), 7.37-7.23 (m, 4H), 7.16-7.08 (m, 2H), 6.80-6.71 (m, 1H), 5.08 (s, 2H), 2.29 (s, 3H); 13C NMR (101 MHz, (CD3)2SO) δ 163.7, 154.5, 142.9, 137.1, 132.8, 132.5, 129.5, 128.6 (2C), 127.5, 126.9 (2C), 126.5, 114.3, 104.8, 69.3, 20.5; vmax 2916, 2848, 1655 (s), 1518, 1256 (s), 1153 (s), 1060, 1016; HRMS-TOF MS ESI+: m/z [M+H]+ calculated for C17H15O3S: 299.0739; found: 299.0742.
4-[(4-Chlorobenzyl)oxy]benzo[b]thiophene-2-carboxylic acid 12c
The general procedure described above was followed using 11c (666 mg, 2.01 mmol), ethanol (30 mL) and lithium hydroxide (363 mg, 15.2 mmol). The pure product was obtained as a white solid (581 mg, 1.82 mmol, 95%). Rf = 0.07, 15% EtOAc/Hex. 1H NMR (400 MHz, (CD3)2 SO) Ô 8.04 (s, 1H), 7.62-7.54 (m, 3H), 7.51-7.43 (m, 3H), 7.04 (d, J = 7.8 Hz, 1H), 5.30 (s, 2H); 13C NMR (101 MHz, (CD3)2SO) δ 163.3, 154.3, 142.8, 135.8, 133.4, 132.5, 129.4, 129.3 (2C), 128.6, 128.5 (2C), 126.0, 115.3, 106.3, 68.6; vmax 3382 (w br), 1576 (s), 1518, 1367 (s), 1151 (s), 1016, 805, 769, 752; HRMS-TOF MS ESI+: m/z [M+H] + calculated for C16H12ClO3S: 319.0199; found: 319.0196.
N1-(7-Chloroquinolin-4-yl)ethane-1,2-diamine 14a
4,7-Dichloroquinoline (1.20 g, 5.15 mmol) and ethane-1,2-diamine (7.00 mL, 6.29 g, 104 mmol) were added to a round-bottom flask. The mixture was heated to 140 °C and stirred under reflux for 1.5 hours and monitored by TLC for completion. On completion, the mixture was cooled to room temperature and the crude mixture poured into a small quantity of cold water. The aqueous mixture was then extracted with ethyl acetate (3 x 20 mL), the organic layers combined and washed with 50 mL of distilled water. The organic layer was collected and dried over anhydrous magnesium sulfate followed by vacuum filtration and evaporating the solvent under reduced pressure to give crude product as an off-white solid. The crude material was purified by adding 3 mL of hexane and heated to boiling, cooled to room temperature and the hexane pipetted off. This process was repeated three times to remove excess amine. The purified product was dried for 24 hours under vacuum and obtained as an off-white solid (1.01 g, 4.55 mmol, 78%). (mp 132-134 °C); 1H NMR (300 MHz, CD3OD) δ 8.33 (d, J5.6 Hz, 1H), 8.08 (d, J 9.0 Hz, 1H), 7.75 (d, J 1.9 Hz, 1H), 7.36 (dd, J 9.0 and 1.9 Hz, 1H), 6.53 (d, J 5.6 Hz, 1H), 3.43 (t, J 6.3 Hz, 2H), 2.97 (t, J 6.3 Hz, 2H); 13C NMR (75 MHz, (CD3)2SO) δ 152.7, 152.4, 149.6, 136.3, 127.5, 126.0, 124.3, 118.7, 99.6, 46.2, 40.8; vmax 3243 (w br), 1582 (s), 1541, 1450, 1426, 1326, 1282, 1161, 1141, 950, 869, 800 (s), 758, 618 (m); HRMS-TOF MS ESI+: m/z [M+H]+ calculated for C11H13ClN3: 222.0799; found: 222.0798. This is a known compound and values correspond well with previously reported values.12
N1-[7-(Trifluoromethyl)quinolin-4-yl]ethane-1,2-diamine 14b
4-Chloro-7-(trifluoromethyl)quinoline (500 mg, 2.16 mmol), ethane-1,2-diamine (2.89 mL, 2.59 g, 43.1 mmol) and toluene (3 mL) were added to a 10 mL microwave vial. The vial was sealed with a vial cap and septum and stirred at 160 °C for 24 hours. On completion, the crude mixture was cooled to room temperature and diluted with 20 mL of distilled water. The aqueous mixture was then extracted with ethyl acetate (3 x 20 mL), the organic layers combined and dried over anhydrous magnesium sulfate followed by vacuum filtration. The collected solution was concentrated under reduced pressure to give the crude product as a white solid. The crude material was purified by adding 3 mL of hexane and heated to boiling point, cooled to room temperature and the hexane pipetted off. This process was repeated three times to remove excess amine and toluene. The purified product was dried for 24 hours under vacuum and obtained as a white solid (448 mg, 1.75 mmol, 81%). (mp 87-89 °C); 1H NMR (300 MHz, CD3OD) δ 8.31 (d, J 5.6 Hz, 1H), 8.16 (d, J 8.8 Hz, 1H), 7.93 (s, 1H), 7.49 (d, J 8.8 Hz, 1H), 6.49 (d, J 5.6 Hz, 1H), 3.38 (t, J 6.2 Hz, 2H), 2.96 (t, J 6.2 Hz, 2H); 13C NMR (75 MHz, (CD3)2SO) δ 143.3, 142.9, 138.6, 123.4 -121.7 (m), 116.9-116.6 (m), 114.7, 112.6, 111.4-111.1 (m), 91.2, 36.0, 30.9; vmax 3271 (w br, N-H, stretch), 2888 (w br), 1593, 1537, 1470, 1457 (m), 1430, 1375, 1322 (s), 1271, 1162, 1125 (w br) 816 (s), 683 (m); HRMS-TOF ESI+: m/z [M+H]+ calculated for C12H13F3N3: 256.1059; found: 256.1062. This is a known compound and values correspond well with previously reported values.13
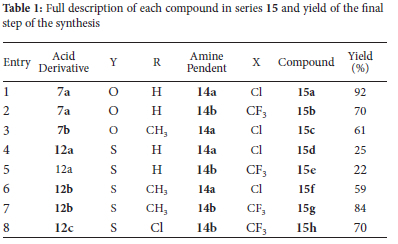
Synthesis of 15a-h
General procedure
Dry DMSO, N,N'-carbonyldiimidazole (CDI, 1.3 equivalents), imidazolium chloride (1.5 equivalents) and one of the respective benzofuran or benzothiophene carboxylic acids (1 equivalent) was added to a microwave vial equipped with a stir bar. The vial was sealed, and the reaction mixture stirred at 55 °C for 1-4 hours and monitored by TLC (15% EtOAc/Hex) for conversion of starting material. Bromocresol green stain was used as visualizing agent to monitor the conversion of the carboxylic acid to the intermediate. On completion the desired quinoline derivative (1.2 equivalents) was added in a single portion to the reaction and the reaction vial resealed. The reaction was stirred for 6-48 hours whilst heating between 60 °C and monitored by TLC (10% MeOH/EtOAc) for the consumption of the intermediate. On completion the reaction mixture was cooled to room temperature and diluted with distilled water until the mixture turned milky in color. The milky aqueous mixture was then extracted with ethyl acetate (4 x 25 mL). The organic layers were combined, washed with distilled water (3 x 25 mL), once with brine followed by drying over anhydrous magnesium sulfate. The solvent was collected by vacuum filtration and evaporated under reduced pressure to afford the crude product as a thick oil or solid. The crude product was purified by various methods and products obtained as white to off-white solids (22-92%).
4-(Benzyloxy)-N-{2-[(7-chloroquinolin-4-yl)amino]ethyl} benzofuran-2-carboxamide 15a
The general procedure was followed using 7a (24.4 mg, 0.0909 mmol), dry DMSO (7 mL), imidazolium chloride (14.5 mg, 0.139 mmol), CDI (19.4 mg, 0.120 mmol) and 14a (26.0 mg, 0.117 mmol). The mixture was stirred at 60 °C for 48 hours and was monitored by TLC for completion. The crude product was obtained as an off-white solid and purified by automated column chromatography (1% triethylamine/ 9% MeOH/ 90% EtOAc) to afford the product as a white solid (39.4 mg, 0.0836 mmol, 92%). (mp 224-226 °C); 1H NMR (600 MHz, (CD3)2SO) δ 8.89 (app. t, J 5.7 Hz, 1H), 8.42 (d, J 5.4 Hz, 1H), 8.21 (d, J 9.0 Hz, 1H), 7.79 (d, J 2.0 Hz, 1H), 7.58 (s, 1H), 7.52 (d, J 7.4 Hz, 2H), 7.49-7.33 (m, 6H), 7.25 (d, J 8.4 Hz, 1H7), 6.96 (d, J 8.1 Hz, 1H), 6.63 (d, J 5.4 Hz, 1H), 5.28 (s, 2H), 3.56-3.51 (m, 2H), 3.48-3.45 (m 2H); 13C NMR (151 MHz, (CD3)2SO) Ô 158.4, 155.4, 152.9, 151.8, 150.0, 149.0, 147.9, 136.7, 133.4, 128.4 (2C), 127.9 (2C), 127.6, 127.5 (2C), 124.1, 123.9, 117.6, 117.4, 106.7, 105.6, 104.7, 98.6, 69.7, 41.8, 37.3; vmax 3363 (w br), 2931, 1656, 1581, 1278 (s), 1225, 1137, 1072, 726; HRMS-TOF ESI+: m/z [M+H]+ calculated for C27H23ClN3O3: 472.1429; found: 472.1425.
4-(Benzyloxy)-N-(2-{[7-(trifluoromethyl)quinolin-4-yl]amino} ethyl)benzofuran-2-carboxamide 15b
The general procedure was followed using 7a (100 mg, 0.373 mmol), dry DMSO (7 mL), imidazolium chloride (56.4 mg, 0.540 mmol), CDI (78.5 mg, 0.484 mmol) and 14b (85.0 mg, 0.333 mmol). The crude product was obtained as an off-white solid and purified by heating in 2 mL of hexane, the hexane cooled and pipetted off. This process was repeated 3 times to afford the product as a white solid (132 mg, 0.262 mmol, 70%). (mp 190-192 °C); 1H NMR (400 MHz, (CD3)2SO) δ 8.73 (app. t, J 5.9 Hz, 1H), 8.46 (d, J 5.4 Hz, 1H), 8.29 (d, J 8.8 Hz, 1H), 8.05 (s, 1H), 7.57-7.49 (m, 2H), 7.45-7.37 (m, 3H), 7.36-7.24 (m, 4H), 7.09 (d, J 8.4 Hz, 1H), 6.95 (s, 1H), 6.76 (d, J 8.0 Hz, 1H), 6.56 (d, J 5.5 Hz, 1H), 5.16 (s, 2H), 3.72-3.64 (m, 2H), 3.53-3.44 (m, 2H); 13C NMR (101 MHz, (CD3)2SO) δ 159.9, 155.4, 152.9, 151.4, 149.7, 146.9, 146.7, 135.9, 130.4 (q, J 32.5 Hz), 128.1 (2C), 127.5, 127.3, 126.9 (2C), 126.1125.1 (m), 124.4, 122.2, 120.2, 119.0, 117.8, 107.6, 104.5, 104.2, 98.8, 69.7, 43.8, 37.6; 19F NMR (282 MHz, (CD3)2SO) δ 62.0 (s, 3F, CF3); vmax 3315 (w br), 1589, 1327 (s), 1267, 1108, 1068, 747;HRMS-TOF ESI+: m/z [M+H]+ calculated for C28H23F3N3O3: 506.1689; found: 506.1698.
N-{2-[(7-chloroquinolin-4-yl)amino]ethyl}-4-[(4-methylbenzyl) oxy]-benzofuran-2-carboxamide 15c
The general procedure was followed using 7b (46.0 mg, 0.163 mmol), dry DMSO (5 mL), imidazolium chloride (19.3 mg, 0.185 mmol), CDI (35.8 mg, 0.221 mmol) and 14a (34.0 mg, 0.153 mmol). The mixture was stirred at 60 °C for 24 hours was monitored by TLC for completion. The crude product was obtained as an off-white solid and purified by automated column chromatography (1/9/90% TEA/MeOH/EtOAc) to afford the product as a white solid (46.9 mg, 0.0971 mmol, 63%); (mp 190-192 °C); 1H NMR (400 MHz, (CD3)2SO) δ 9.08 (app. t, J 5.7 Hz, 1H), 8.41 (d, J 4.9 Hz, 1H), 8.35 (d, J 9.1 Hz, 1H), 7.79 (br s, 1H), 7.71-7.65 (m, 1H), 7.64 (s, 1H), 7.46-7.41 (m, 1H), 7.40-7.33 (m, 3H), 7.24-7.18 (m, 3H), 6.92 (d, J 8.0 Hz, 1H), 6.62 (d, J 5.3 Hz, 1H), 5.22 (s, 2H), 3.62-3.53 (m, 2H), 3.52-3.44 (m, 2H), 2.30 (s, 3H); 13C NMR (101 MHz, (CD3)2SO) δ 158.4, 155.4, 153.0, 151.7, 150.1, 148.9, 147.8, 137.1, 133.7, 133.4, 129.0 (2C), 127.8, 127.7 (2C), 127.3, 124.3, 124.1, 117.6, 117.4, 106.7, 105.6, 104.6, 98.6, 69.6, 41.9, 37.3, 20.7; vmax 3328 (w br), 2922, 1652, 1578, 1269 (s), 1074, 795, 727; HRMS-TOF ESI-: m/z [M-H]- calculated for C28H25ClN3O3: 484.1427; found: 484.1430
4-(Benzyloxy)-N-{2-[(7-chloroquinolin-4-yl)amino]ethyl} benzo[b]thiophene-2-carboxamide 15d
The general procedure was followed using 12a (101 mg, 0.355 mmol), dry DMSO (5 mL), imidazolium chloride (58.3 mg, 0.558 mmol), CDI (78.1 mg, 0.482 mmol) and 14a (92.3 mg, 0.416 mmol). The crude product was obtained as an off-white solid and purified by heating in 2 mL of hexane, the hexane cooled and pipetted off. This process was repeated 3 times to afford the product as a white solid (45.0 mg, 0.0924 mmol, 26%). (mp 224-226 °C); 1H NMR (600 MHz, (CD3)2SO) Ô 9.19 (app. t, J 5.6 Hz, 1H), 8.39 (br s, 1H), 8.34 (d, J 9.1 Hz, 1H), 8.22 (s, 1H), 7.80-7.74 (m, 2H), 7.54 (d, J 7.1 Hz, 2H), 7.50 (d, J 8.1 Hz, 1H), 7.42-7.30 (m, 5H), 6.99 (d, J7.9 Hz, 1H, H-5), 6.61 (d, J 5.4 Hz, 1H), 5.25 (s, 2H), 3.58-3.53 (m, 2H), 3.52-3.46 (m, 2H); 13C NMR (151 MHz, (CD3)2SO) δ 165.1, 157.5, 154.7, 153.1, 144.8, 141.1, 139.6, 136.5, 133.1, 131.4 (2C), 131.0, 130.9 (2C), 130.5, 130.4, 127.1, 126.9, 124.2, 120.5, 117.9, 108.7, 101.5, 72.7, 45.0, 40.9; vmax 3345 (w br), 2972, 1640, 1536, 1227 (s), 1023, 734, 696;HRMS-TOF ESI+: m/z [M+H]+ calculated for C27H23ClN3O2S: 488.1199; found: 488.1200.
4-(Benzyloxy)-N-(2-{[7-(trifluoromethyl)quinolin-4-yl]amino} ethyl)benzo[b]thiophene -2-carboxamide 15e
The general procedure was followed using 12a (150 mg, 0.527 mmol), dry DMSO (7 mL), imidazolium chloride (84.0 mg, 0.804 mmol), CDI (108 mg, 0.666 mmol) and 14b (134 mg, 0.524 mmol). The crude product was obtained as a white solid and purified by heating in 2 mL of hexane, the hexane cooled and pipetted off. This process was repeated 3 times to afford the product as a white solid (61.4 mg, 0.118 mmol, 22%). (mp 218-220 °C); 1H NMR (600 MHz, (CD3)2SO) δ 8.38 (d, J 5.6 Hz, 1H), 8.23 (d, J 8.8 Hz, 1H), 8.04 (s, 1H), 7.99 (s, 1H), 7.56 (d, J 8.8 Hz, 1H), 7.47-7.39 (m, 3H), 7.34-7.24 (m, 4H), 6.91 (d, J 7.9 Hz, 1H), 6.66 (d, J 5.6 Hz, 1H), 5.14 (s, 2H), 3.61-3.56 (m, 2H), 3.543.48 (m, 2H); 13C NMR (151 MHz, CD3OD) Ô 164.1, 155.8, 152.7, 151.7, 147.9, 143.2, 138.6, 137.8, 131.62-130.86 (q, J 32.2 Hz, 1C, CF3), 131.2, 129.6, 129.3 (2C), 128.9, 128.8 (2C), 126.3-126.1 (m), 124.2, 122.5, 121.7, 120.3-120.1 (m), 115.7, 106.6, , 100.5, 70.8, 43.2, 38.9; vmax 3344 (w br), 2943, 1544, 1327 (s), 1261, 1131, 735; HRMS-TOF ESI+: m/z [M+H]+ calculated for C28H23F3N3O2S: 522.1464; found: 522.1463.
N -{2-[(7-Chloroquinolin-4-yl)amino]ethyl}-4-[(4-methylbenzyl) oxy]benzo[b]thiophene -carboxamide 15f
The general procedure was followed using 12b (153 mg, 0.512 mmol), dry DMSO (7 mL), imidazolium chloride (85 mg, 0.81 mmol), CDI (107 mg, 0.660 mmol) and 14a (134 mg, 0.604 mmol). The crude product was obtained as a thick oil and purified by automated step gradient column chromatography (50% EtOAc/Hex then 10% MeOH/ EtOAc) to afford the product as a white solid (149 mg, 0.297 mmol, 58%). (mp 147-149 °C); 1H NMR (400 MHz, (CD3)2SO) δ 9.02 (app. t, J 5.6 Hz, 1H), 8.40 (d, J 5.4 Hz, 1H), 8.21-8.09 (m, 2H), 7.78 (d, J 2.2 Hz, 1H), 7.47 (d, J 8.1 Hz, 1H), 7.43-7.33 (m, 4H), 7.19 (d, J 7.9 Hz, 2H), 6.93 (d, J7.9 Hz, 1H), 6.55 (d, J 5.5 Hz, 1H), 5.15 (s, 2H), 3.63-3.55 (m, 2H), 3.53-3.45 (m, 2H), 2.32 (s, 3H); 13C NMR (101 MHz, (CD3)2SO) δ 162.0, 154.4, 151.6, 149.9, 148.9, 141.7, 138.2, 137.2, 133.4, 133.3, 129.9, 128.8 (2C), 127.9 (2C), 127.4, 127.2, 123.9, 123.5, 121.1, 117.3, 114.6, 105.4, 98.3, 69.5, 42.0, 37.8, 20.7; vmax 3338 (w br), 2928, 1557, 1257 (s), 1021, 799; HRMS-TOF ESI+: m/z [M+H] + calculated for C28H25ClN3O2S: 502.1357; found: 502.1356.
N-(2-{[7-(Trifluoromethyl)quinolin-4-yl]amino}ethyl)-4-[(4-methylbenzyl)oxy]benzofuran-2-carboxamide 15g
The general procedure was followed using 12b (150 mg, 0.503 mmol), dry DMSO (7 mL), imidazolium chloride (77.4 mg, 0.740 mmol), CDI (104 mg, 0.641 mmol) and 14b (154 mg, 0.603 mmol). The crude product was obtained as a thick oil and purified by automated step gradient column chromatography (50% EtOAc/Hex then 10% MeOH/ EtOAc) to afford the product as a white solid (230 mg, 0.429 mmol, 85%). (mp 228-230 °C); 1H NMR (600 MHz, (CD3)2SO) Ô 9.04 (app. t, J 5.0 Hz, 1H), 8.52 (d, J 5.2 Hz, 1H), 8.42 (d, J 8.8 Hz, 1H), 8.13 (s, 1H), 8.09 (s, 1H), 7.69 (d, J 7.8 Hz, 1H), 7.67-7.63 (m, J 5.0 Hz, 1H), 7.56 (d, J 8.1 Hz, 1H), 7.45-7.36 (m, 3H), 7.22 (d, J 7.7 Hz, 2H), 7.03 (d, J 7.9 Hz, 1H), 6.74 (d, J 5.4 Hz, 1H), 5.21 (s, 2H), 3.59-3.47 (m, 4H), 2.31 (s, 3H); 13C NMR (151 MHz, (CD3)2SO) Ô 161.9, 154.4, 152.2, 149.9, 147.4, 141.6, 138.4, 137.3, 133.6, 130.0, 129.1 (2C), 129.0, 128.1 (2C), 127.6, 126.2, 123.8, 121.1, 120.9, 118.9, 114.9, 105.9, 99.9, 69.5, 41.8, 39.5, 37.7, 20.7; vmax 3348 (w br), 2928, 1639, 1545, 1325 (s), 1123, 901, 801; HRMS-TOF MS ESI+: m/z [M+H]+ calculated for C29H25F3N3O2S: 536.1620; found: 536.1642.
4-[(4-Chlorobenzyl)oxy]-N-(2-{[7-(trifluoromethyl)quinolin-4-yl] amino}ethyl)benzo[b]thiophene-carboxamide 15h
The general procedure was followed using 12c (101 mg, 0.317 mmol), dry DMSO (7 mL), imidazolium chloride (79.8 mg, 0.763 mmol), CDI (166 mg, 1.02 mmol) and 14b (97.6 mg, 0.382 mmol). The crude product was obtained as an off-white solid and purified by automated column chromatography (5/45/50% TEA/EtOAc/Hex) to afford the product as a white solid (121 mg, 0.217 mmol, 69%). (mp 230230 °C); 1H NMR (400 MHz, (CD3)2SO) δ 9.11-9.00 (m, 1H), 8.51 (d, J 4.6 Hz, 1H), 8.40 (d, J 8.0 Hz 1H), 8.15 (s, 1H), 8.08 (s, 1H, H-23), 7.70-7.60 (m, 2H), 7.57-7.49 (m, 3H), 7.45 (d, J 8.1 Hz, 2H), 7.37 (t, J 7.9 Hz, 1H), 6.98 (d, J 7.8 Hz, 1H), 6.71 (d, J 5.2 Hz, 1H), 5.23 (s, 2H), 3.61-3.46 (m, 4H); 13C NMR (101 MHz, (CD3)2SO) δ 161.9, 154.2, 152.3, 149.9, 147.4, 141.7, 138.5, 135.7, 132.7, 129.9, 129.7 (2C), 128.5 (2C), 127.5, 126.3, 125.5, 123.8, 122.8, 121.1, 120.9, 118.9, 115.1, 105.9, 99.7, 68.7, 41.8, 37.7; vmax 3346 (w br), 1640, 1548, 1325 (s), 1122, 808; HRMS-TOF ESI+: m/z [M+H]+ calculated for C28H22ClF3N3O2S: 556.1074; found: 556.1073.
2-[(7-Chloroquinolin-4-yl)amino]ethanol 16a
4,7-Dichloroquinoline (1.00 g, 5.05 mmol), 2-aminoethanol (6.11 mL, 6.17 g, 100 mmol), n-butanol (1.5 mL) and toluene (10 mL) were added to a 25 mL microwave vial equipped with a stir bar. The vial was sealed with a crimp cap, heated to 160 °C and stirred for 24 hours. On completion, the reaction was cooled to room temperature and the reaction mixture diluted with distilled water (25 mL) and ethyl acetate (10 mL) added. Semi-pure product precipitated out of the aqueous-organic mixture and was collected by filtration followed by washing with a single portion of ethyl acetate (10 mL). The product was collected and dried under vacuum for 24 hours to remove residual butanol and amine with the final product obtained as an off-white solid (923 mg, 4.14 mmol, 82%). (mp 216-218 °C); 763; 1H NMR (400 MHz, (CD3)2SO) δ 8.38 (d, J 5.3 Hz, 1H), 8.26 (d, J 9.0 Hz 1H), 7.78 (d, J 2.1 Hz, 1H), 7.44 (dd, J 8.9 Hz and 2.1 Hz, 1H), 7.30-7.23 (m, 1H), 6.49 (d, J 5.4 Hz, 1H), 4.86 (broad s, 1H), 3.66 (t, J 5.7 Hz, 2H), 3.423.30 (m, 2H); 13C NMR (101 MHz, (CD3)2SO) Ô 151.8, 150.2, 149.0, 133.3, 127.4, 124.1, 124.0, 117.4, 98.7, 58.7, 45.1; vmax 3306 (w br), 2817, 1614, 1580 (s), 1537, 1431, 1333 (m), 1220, 1138, 1062 (s), 924, 883, 867, 819, 800 (s), HRMS-TOF ESI+: m/z [M+H]+ calculated for C11H12ClN2O: 223.0639; found: 223.0638. This is a known compound and values correspond well with previously reported values.14
2-{[7-(Trifluoromethyl)quinolin-4-yl]amino}ethanol 16b
4-Chloro-7-trimethylquinoline (502 mg, 2.16 mmol), 2-aminoethanol (2.61 mL, 2.64 g, 43.1 mmol), n-butanol (1.5 mL) and toluene (10 mL) were added to a 25 mL microwave vial equipped with a stir bar. The vial was sealed with a crimp cap, heated to 160 °C and stirred for 24 hours. On completion, the reaction was cooled to room temperature and the reaction mixture diluted with distilled water (25 mL). The aqueous mixture was extracted with ethyl acetate (3 x 25 mL) and the organic layers combined, dried over anhydrous magnesium sulfate and the solution collected by vacuum filtration. The solvent was evaporated under reduced pressure to afford the product as a crude off-white solid. The crude material was purified by adding 3 mL of hexane, heated to boiling point, cooled to room temperature and the hexane pipetted off. This process was repeated three times to remove excess amine, butanol and toluene. The purified product was dried for 24 hours under vacuum and obtained as a white solid (348 mg, 1.35 mmol, 63%). (mp 181183 °C); 1H NMR (400 MHz, (CD3)2SO) δ 8.49 (d, J 5.4 Hz, 1H), 8.47 (d, J 8.8 Hz, 1H), 8.08 (s, 1H), 7.68 (dd, J 8.7 Hz, J 1.4 Hz, 1H), 7.45-7.36 (m, 1H), 6.61 (d, J 5.4 Hz, 1H), 4.87 (broad s, 1H), 3.73-3.64 (m, 2H), 3.43-3.33 (m, 2H); 13C NMR (101 MHz, (CD3)2SO) Ô 152.2, 150.1, 147.4, 129.4-128.4, (q, J 31.9 Hz, 1C), 126.4-126.2 (m, 1C), 123.9, 120.9, 118.8-118.7 (m, 1C) 99.7, 58.7, 45.2; vmax 3116 (w br), 2874, 1596 (m), 1552, 1474, 1376, 1323 (s), 1284, 1206, 1154, 1142, 1106 (s), 1067, 1026, 921, 895, 811(m), 736, 680; HRMS-TOF ESI+: m/z [M+H]+ calculated for C12H12F3N2O: 257.0899; found: 257.0902. This is a known compound and values correspond well with previously reported values.15
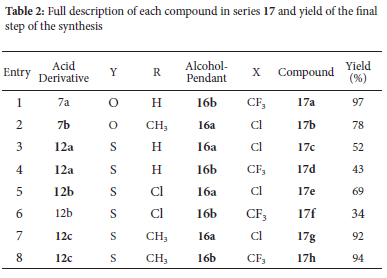
Synthesis of 17a-h
General procedure
One of the respective benzofuran 7a-b or benzothiophene acids 12a-c (1 equivalent), dry DCM (5-10 mL), N,N'-diisopropylcarbodiimide (DIC, 1.5 equivalents), 4-dimethylaminopyridine (DMAP, 0.3 equivalents) and the desired quinoline alcohol derivative (0.9 equivalents) were added to a microwave vial equipped with a stir bar, vial cap and rubber septum. The reaction vial was sealed, and the reaction mixture stirred at 60 °C for 24-48 hours and monitored by TLC (50% EtOAc/Hex) for conversion of starting material. On completion the reaction mixture was cooled to room temperature, diluted with distilled water and then a small amount of acetone added. This mixture was stirred for 5 minutes before transferring to a separating funnel and extracted with EtOAc (3 x 20 mL). The organic layers were combined and washed with distilled water (3 x 20 mL) and once with brine. The organic layers were dried over anhydrous magnesium sulfate, the solvent was collected by vacuum filtration and evaporated under reduced pressure to afford the crude product as a thick oil. The crude product was purified by various methods depending on the product.
2-{[7-(Trifluoromethyl)quinolin-4-yl]amino}ethyl-4-(benzyloxy) benzofuran-2-carboxylate 17a
The general procedure was followed using 7a (75 mg, 0.28 mmol), dry DCM (5 mL), DMAP (12.7 mg, 0.104 mmol), DIC (0.07 mL, 50 mg, 0.4 mmol) and 16b (72.2 mg, 0.282 mmol). The crude product was obtained as a pale yellow solid and purified by automated step gradient column chromatography (100% Hex then 15% EtOAc/Hex then 100% EtOAc) to afford the product as a white solid (138 mg, 0.273 mmol, 97%). (mp 184-186 °C); 1H NMR (400 MHz, (CD3)2SO) δ 8.38 (d, J 5.3 Hz, 1H), 8.22 (d, J 9.1 Hz, 1H), 8.11 (s, 1H), 7.74 (d, J 2.0 Hz, 1H), 7.52-7.46 (m, 4H), 7.42-7.29 (m, 5H), 6.96 (d, J 7.9 Hz, 1H), 6.60-6.57 (m, 1H), 5.25 (s, 2H), 4.54 (t, J 5.7 Hz, 2H), 3.71 (m, 2H); 13C NMR (101 MHz, (CD3)2SO) δ 158.5, 156.9, 156.1, 153.1, 151.6, 149.6, 147.2, 143.3, 135.8, 130.0, 128.9 (q, J 32.2 Hz, CF3), 128.4, 128.0 (2C), 127.6, 127.0 (2C), 126.3-126.0 (m), 124.9, 122.9, 122.2, 118.8-118.5 (m), 117.3, 111.6, 104.7, 104.4, 99.2, 69.6, 62.5, 41.0; vmax 3339 (w br), 2967, 1719 (s), 1573, 1244, 1161, 1129, 1070, 754; HRMS-TOF ESI+: m/z [M+H]+ calculated for C28H22F3N2O4: 507.1532; found: 507.1533.
2-[(7-Chloroquinolin-4-yl)amino]ethyl-4-[(4-methylbenzyl)oxy]-benzofuran-2-carboxylate 17b
The general procedure described was followed using 7b (179 mg, 0.634 mmol), dry DCM (10 mL), DMAP (25.6 mg, 0.210 mmol), DIC (0.15 mL, 120 mg, 0.95 mmol) and 16a (127 mg, 0.570 mmol). Reaction temperature was increased from 60 °C to 90 °C after 2 hours and then stirred for a further 18 hours due to slow conversion of starting material. The crude product was obtained as a pale yellow solid and purified by automated step gradient column chromatography (15% EtOAc/Hex, 50% EtOAc/Hex then 100% EtOAc) to afford the product as a white solid (260 mg, 0.535 mmol, 94%). 1H NMR (600 MHz, CDCl3) δ 8.40 (d, J = 4.4 Hz, 1H), 8.23 (d, J 9.1 Hz, 1H), 7.77 (s, 1H), 7.63 (s, 1H), 7.52-7.46 (m, 1H), 7.36-7.27 (m, 3H), 7.19-7.11 (m, 3H), 6.80 (d, J 8.1 Hz, 1H), 6.53 (d, J 5.4 Hz, 1H), 5.12 (s, 2H), 4.53 (t, J 5.6 Hz, 2H), 3.76-3.67 (m, 3H), 2.30 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 158.4, 157.0, 156.1, 153.2, 151.3, 149.9, 148.8, 143.5, 137.0, 133.6, 133.1, 128.8 (2C), 128.6, 127.3 (2C), 123.9, 123.6, 117.3, 111.5, 105.0, 104.4, 98.4, 69.6, 62.5, 41.0, 39.5, 20.6; vmax 3338 (w br, N-H), 2967, 1720 (s), 1612, 1569, 1247, 1166, 753; HRMS-TOF ESI+: m/z [M+H] + calculated for C28H24ClN2O4: 487.1425; found: 487.1436.
2-[(7-Chloroquinolin-4-yl)amino]ethyl-4-(benzyloxy)benzo[b] thiophene-2-carboxylate 17c
The general procedure was followed using 12a (100 mg, 0.352 mmol), dry DCM (5 mL), DMAP (10.0 mg, 0.0819 mmol), DIC (0.08 mL, 70 mg, 0.5 mmol) and 16a (85.6 mg, 0.384 mmol). The crude product was obtained as a yellow solid and purified by automated step gradient column chromatography (30% EtOAc/Hex then 100% EtOAc) to afford the product as a white solid (91.4 mg, 0.187 mmol, 53%). (mp 190-192 °C); 1H NMR (400 MHz, (CD3)2SO) δ 8.42 (d, J 5.2 Hz, 1H), 8.26 (d, J 9.1 Hz, 1H), 8.10 (s, 1H), 7.80 (d, J 2.0 Hz, 1H), 7.59-7.55 (m), 7.51 (d, J 7.1 Hz, 2H), 7.48-7.32 (m, 5H), 7.05 (d, J 7.9 Hz, 1H), 6.67 (d, J 5.5 Hz, 1H), 5.29 (s, 2H), 4.54 (t, J 5.4 Hz, 2H), 3.77-3.69 (m, 2H); 13C NMR (101 MHz, (CD3)2SO) Ô 152.4, 157.2, 155.1, 152.1, 150.5, 149.2, 143.2, 137.0, 134.0, 131.8, 129.7, 129.4, 128.9 (2C), 128.4, 128.0 (2C), 127.8, 127.7, 124.7, 124.4, 115.6, 106.9, 99.4, 70.0, 63.8, 23.7; vmax 3338 (w br), 2966, 1720 (s), 1567, 1235, 1018, 746;HRMS-TOF ESI+: m/z [M+H]+ calculated for C27H22ClN2O3S: 489.1040; found: 489.1040.
2-{[7-(Trifluoromethyl)quinolin-4-yl]amino}ethyl-4-(benzyloxy) benzo[b]thiophene -2-carboxylate 17d
The general procedure was followed using 12a (200 mg, 0.703 mmol), dry D CM (2 x 5 mL), DMAP (17.2 mg, 0.141 mmol), DIC (0.12 mL, 97.6 mg, 0.77 mmol), and 16b (161 mg, 0.628 mmol). The crude product was collected by filtration and washed with water (3 x 10 mL) then hexane (1 x 15 mL) to afford the product as a white solid (156 mg, 0.299 mmol, 48%). (mp 196-198 °C); 1H NMR (400 MHz, (CD3)2SO) Ô 8.50 (d, J 5.4 Hz, 1H), 8.43 (d, J 8.8 Hz, 1H), 8.12-8.04 (m, 2H), 7.657.59 (m, 2H), 7.57 (d, J 8.2 Hz, 1H), 7.53-7.29 (m, 6H), 7.03 (d, J 7.9 Hz, 1H), 6.77 (d, J 5.4 Hz, 1H), 5.28 (s, 2H), 4.60-4.50 (m, 2H), 3.783.69 (m, 2H); 13C NMR (101 MHz, (CD3)2SO) δ 161.9, 156.9, 154.7, 149.9, 147.4, 136.6, 131.4, 129.3, 119.0-118.5 (m), 115.2, 106.4, 100.0, 69.5, 63.4, 40.6; 19F NMR (282 MHz, (CD3)2SO) δ 60.8 (s, 3F, CF3); vmax 3338 (w br, N-H, stretch, amine), 2967, 1721 (s, C=O, stretch, ester), 1568, 1328, 1237, 1127, 1020, 747; HRMS-TOF ESI+: m/z [M+H]+ calculated for C28H22F3N2O3S: 523.1304; found: 523.1315.
2-[(7-Chloroquinolin-4-yl)amino]ethyl-4-[(4-chlorobenzyl)oxy] benzo[b]thiophene-carboxylate 17e
The general procedure was followed using 12c (100 mg, 0.314 mmol), dry D CM (7 mL), DMAP (11.0 mg, 0.0900 mmol), DIC (0.07 mL, 60 mg, 0.5 mmol) and 16a (63.8 mg, 0.287 mmol). The crude product was obtained as a yellow solid and purified by automated column chromatography (50% EtOAc/Hex) to afford the product as a white solid (114 mg, 0.218 mmol, 76%). (mp 160-162 °C); 1H NMR (400 MHz, (CD3)2SO) δ 8.39 (d, J 5.4 Hz, 1H), 8.23 (d, J 9.1 Hz, 1H), 8.08 (s, 1H), 7.77 (d, J 2.1 Hz, 1H), 7.57 (t, J 7.7 Hz, 1H, H-7), 7.55-7.38 (m, 7H), 7.02 (d, J 7.9 Hz, 1H), 6.65 (d, J 5.4 Hz, 1H), 5.28 (s, 2H), 4.52 (t, J 5.3 Hz, 2H), 3.76-3.65 (m, 2H); 13C NMR (101 MHz, (CD3)2SO) δ 162.3, 157.2, 152.2, 150.5, 149.4, 143.2, 136.1, 133.9, 133.0, 131.9, 129.9, 129.7 (2C), 129.4, 128.9 (2C), 127.9, 127.2, 124.6, 124.4, 117.8, 115.7, 106.9, 99.4, 69.1, 63.8, 41.5; vmax 3636 (w br), 2919, 1707 (s), 1583, 1348, 1245, 1015, 807, 747; HRMS-TOF ESI+: m/z [M+H]+ calculated for C27H21Cl2N2O3S: 523.0651; found: 523.0650.
2-{[7-(Trifluoromethyl)quinolin-4-yl]amino}ethyl-4-[(4-chlorobenzyl)oxy]benzo[b]thiophene-carboxylate 17f
The general procedure was followed using 12c (51 mg, 0.160 mmol), dry DCM (7 mL), DMAP (5.9 mg, 0.048 mmol), DIC (0.04 mL, 30 mg, 0.2 mmol) and 16b (36.0 mg, 0.140 mmol). The crude product was obtained as a yellow solid and purified by automated column chromatography (50% EtOAc/Hex) to afford the product as a white solid (31 mg, 0.056 mmol, 40%). 1H NMR (600 MHz, (CD3)2SO) δ 8.52 (d, J 4.7 Hz, 1H), 8.46 (d, J 8.7 Hz, 1H), 8.12-8.06 (m, 2H), 7.73-7.64 (m, 1H), 7.59 (d, J 8.1 Hz), 7.53 (d, J 8.1 Hz, 2H), 7.49-7.41 (m, 3H), 7.03 (d, J 7.9 Hz, 1H), 6.79 (d, J 5.3 Hz, 1H), 5.29 (s, 2H), 4.59-4.53 (m, 2H), 3.81-3.73 (m, 3H). 13C NMR (101 MHz, (CD3)2SO) δ 167.2, 161.9, 154.5, 153.7, 152.1, 149.9, 142.8, 137.0, 135.6, 132.5, 131.5, 129.4, 129.2 (2C), 128.9, 128.5 (2C), 126.8, 126.5-126.3 (m), 123.8, 120.8, 119.0, 115.3, 100.0, 68.7, 63.4, 42.3; vmax 3266 (w br), 2916, 1705 (s), 1550, 1327, 1240, 1111, 809, 747; HRMS-TOF ESI+: m/z [M+H]+ calculated for C28H21ClF3N2O3S: 557.0914; found: 557.0916.
2-[(7-Chloroquinolin-4-yl)amino]ethyl-4-[(4-methylbenzyl)oxy] benzo[b]thiophene -carboxylate 17g
The general procedure was followed using 12b (103 mg, 0.345 mmol), dry D CM (7 mL), DMAP (12.5 mg, 0.102 mmol), DIC (0.08 mL, 60 mg, 0.5 mmol) and 16a (67.7 mg, 0.304 mmol). The crude product was obtained as a yellow solid and purified by automated step gradient column chromatography (30% EtOAc/Hex then 100% EtOAc) to afford the product as a white solid (135 mg, 0.269 mmol, 88%). (mp 216-218 °C); 1H NMR (400 MHz, (CD3)2SO) δ 8.41 (d, J 5.3 Hz, 1H),
8.25 (d, J 9.1 Hz, 1H), 8.07 (s, 1H), 7.80 (d, J 2.0 Hz, 1H), 7.61-7.50 (m, 2H), 7.48-7.34 (m, 4H), 7.19 (d, J 7.7 Hz, 2H), 7.03 (d, J 7.9 Hz, 1H), 6.66 (d, J 5.4 Hz, 1H), 5.23 (s, 2H), 4.53 (t, J 5.2 Hz, 2H), 3.773.66 (m, J 5.3 Hz, 2H), 2.30 (s, 3H); 13C NMR (101 MHz, (CD3)2SO) Ô 152.0, 154.7, 151.9, 150.0, 149.0, 142.8, 137.3, 133.6, 133.5, 131.4, 129.3, 129.1 (2C), 127.8 (2C), 127.5, 126.8, 124.2, 124.0, 117.4, 115.1, 106.5, 99.0, 69.5, 63.4, 41.1, 39.5, 20.8; vmax 3217 (w br), 2972, 1719 (s), 1562, 1242, 1018, 810, 748; HRMS-TOF ESI+: m/z [M+H]+ calculated for C28H24ClN2O3S: 503.1197; found: 503.1196.
2-{[7-(Trifluoromethyl)quinolin-4-yl]amino}ethyl-4-[(4-methylbenzyl)oxy]benzofuran-2-carboxylate 17h
The general procedure was followed using 12b (103 mg, 0.345 mmol), dry DCM (7 mL), DMAP (12.3 mg, 0.101 mmol), DIC (0.08 mL, 60 mg, 0.50 mmol) and 16b (77.2 mg, 0.301 mmol). The crude product was obtained as a yellow solid and purified by automated step gradient column chromatography (30% EtOAc/Hex then 100% EtOAc) to afford the product as a white solid (149 mg, 0.278 mmol, 93%). mp 236-238 °C;; 1H NMR (400 MHz, (CD3)2SO) δ 8.49 (d, J 5.2 Hz, 1H), 8.43 (d, J 8.8 Hz, 1H), 8.09 (s, 1H), 8.05 (s, 1H), 7.67-7.59 (m, 1H), 7.55 (d, J 8.8 Hz, 1H), 7.47 (d, J 8.0 Hz, 1H), 7.42-7.32 (m, 3H), 7.16 (d, J 7.7 Hz, 2H), 6.95 (d, J 7.8 Hz, 1H), 6.69 (d, J 5.3 Hz, 1H), 5.19 (s, 2H), 4.55 (t, J 5.5 Hz, 2H), 3.74 (d, J 5.6 Hz, 2H), 3.60 (t, J 6.2 Hz, 1H), 2.30 (s, 3H); 13C NMR (101 MHz, (CD3)2SO) δ 161.8, 154.6, 151.8, 149.8, 147.3, 142.7, 137.0, 133.2, 131.1, 129.3, 129.2, 128.8 (2C), 128.5, 127.4 (2C), 126.8, 126.2, 123.5, 120.7, 118.6, 114.6, 105.8, 99.5, 69.4, 62.9, 41.0, 20.8; vmax 3228 (w br), 2982, 1719 (s), 1567, 1326, 1234, 1123, 1016, 815, 747; HRMS-TOF ESI+: m/z [M+H]+ calculated for C29H24F3N2O3S: 537.1460; found: 537.1460.
Assays
In vitro Antiplasmodial activity
Screening conducted externally by Drug Discovery and Development Centre (H3D) at Cape Town University. Test samples were screened for in vitro antiplasmodial activity against a chloroquine sensitive (CQS) strain (NF54) strain of the malaria parasite P. falciparum. Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified version of the method by Trager and Jensen16 Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler and Hinrichs.17 Test samples were tested in triplicate on two separate occasions with further dilutions being prepared in complete medium on the day of the experiment. Samples were tested as a suspension if not completely dissolved. Chloroquine and artesunate were used as the reference drugs. A full dose-response was performed starting at a concentration of 3000 nM solution in DMSO, which was then serially diluted 2-fold in complete medium to give 10 concentrations: with the lowest concentration being approximately 6 nM. The same dilution techniques were used for all samples. Reference drugs were tested at a starting concentration of 1000 ng/m. The highest concentration of solvent to which the parasites were exposed has no measurable effect on the parasite viability and data was not provided.
Toxicity
The test samples were prepared to a 10mmol/L stock solution in 100% DMSO. Samples were tested as a suspension if not completely dissolved. Further dilutions were prepared in growth media on the day of the experiment. The standard cytotoxic compound emetine was used as the reference drug in all experiments. A dual-point dose-response evaluation was performed for the test compounds in a 96-well plate to determine the growth inhibition at each concentration. The highest concentration of solvent to which the cells were exposed
was <0.1% and has no measurable effect on viability (data not shown). The assay plate was incubated at 37°C for 48h under 5% CO2. After 44h, a volume of 25μL of MTT solution was added to all the wells in the assay plate. Plates were reincubated for a further 4h to allow the dye to reduce. Crystals were dissolved using 100|L of dimethyl sulfoxide and then absorbance of each well was quantified using a spectrophotometer at 540nM wavelength.18
The remaining population of cells at each concentration of the test compound was determined by comparing the absorbance of each well to the absorbance of a well containing the drug-free control. Survival was plotted against concentration and the amount of inhibition was calculated at each concentration via regression analysis.
β-hematin
This was determined in a Nonidet P-40 (NP-40) detergent system according to a previously reported method19,20 in 96-well plates. IC50 values were determined from triplicate measurements, together with the standard error of the mean (SEM).
SUPPLEMENTARY MATERIAL
Supplementary information for this article is provided in the online supplement.
ORCID IDs
Jonathan Hay: https://orcid.org/0000-0003-2366-2659
Katherine A de Villiers: https://orcid.org/0000-0002-2932-395X
Dale Taylor: https://orcid.org/0000-0001-7402-0563
Tania Olivier: https://orcid.org/0000-0001-9022-8671
Willem AL van Otterlo: https://orcid.org/0000-0002-3300-6463
Margaret AL Blackie: https://orcid.org/0000-0002-2465-4987
REFERENCES
1. Jacobs L, de Kock C, Taylor D, Pelly SC, Blackie MAL. Synthesis of five libraries of 6,5-fused heterocycles to establish the importance of the heterocyclic core for antiplasmodial activity. Bioorg Med Chem. 2018;26(21):5730-5741. https://doi.org/10.1016/j.bmc.2018.10.029. [ Links ]
2. Liu M, Wilairat P, Go M-L. Antimalarial alkoxylated and hydroxylated chalones: structure-activity relationship analysis. J Med Chem. 2001;44(25):4443-4452. https://doi.org/10.1021/jm0101747. [ Links ]
3. Konstantinovic J, Videnovic M, Srbljanovic J, Djurkovic-Djakovic O, Bogojevic K, Sciotti R, Solaja B. Antimalarials with Benzothiophene moieties as aminoquinoline partners. molecules. 2017;22(3):343. https://doi.org/10.3390/molecules22030343. [ Links ]
4. Nevagi RJ, Dighe SN, Dighe SN. Biological and medicinal significance of benzofuran. Eur J Med Chem. 2015;97:561-581. https://doi.org/10.1016/j.ejmech.2014.10.085. [ Links ]
5. Karagöz AÇ, Leidenberger M, Hahn F, Hampel F, Friedrich O, Marschall M, Kappes B, Tsogoeva SB. Synthesis of new betulinic acid/betulin-derived dimers and hybrids with potent antimalarial and antiviral activities. Bioorg Med Chem. 2019;27(1):110-115. https://doi.org/10.1016/j.bmc.2018.11.018. [ Links ]
6. Kaschula CH, Egan TJ, Hunter R, Basilico N, Parapini S, Taramelli D, Pasini E, Monti D. Structure-activity relationships in 4-aminoquinoline antiplasmodials. The role of the group at the 7-position. J Med Chem. 2002;45(16):3531-3539. https://doi.org/10.1021/jm020858u. [ Links ]
7. Masubuchi M, Kawasaki K-I, Ebiike H, Ikeda Y, Tsujii S, Sogabe S, Fujii T, Sakata K, Shiratori Y, Aoki Y, et al. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N -myristoyltransferase. Part 1. Bioorg Med Chem Lett. 2001;11(14):1833-1837. https://doi.org/10.1016/S0960-894X(01)00319-5. [ Links ]
8. Dayal B, Salen G, Toome B, Tint G, Shefer S, Padia J. Lithium hydroxide/aqueous methanol: mild reagent for the hydrolysis of bile acid methyl esters. Steroids. 1990;55(5):233-237. https://doi.org/10.1016/0039-128X(90)90021-3. [ Links ]
9. Simonetti SO, Larghi EL, Kaufman TS. A convenient approach to an advanced intermediate toward the naturally occurring, bioactive 6-substituted 5-hydroxy-4-aryl-1H-quinolin-2-ones. Org Biomol Chem. 2016;14(9):2625-2636. https://doi.org/10.1039/C5OB0268OT. [ Links ]
10. Woodman EK, Chaffey JG, Hopes PA, Hose DR, Gilday JP. N,N '-Carbonyldiimidazole-mediated amide coupling: significant rate enhancement achieved by acid catalysis with imidazole-HCl. Org Process Res Dev. 2009;13(1):106-113. https://doi.org/10.1021/op800226b. [ Links ]
11. Hanessian S, Pan J, Carnell A, Bouchard H, Lesage L. Total synthesis of (-)-reserpine using the chiron approach. J Org Chem. 1997;62(3):465-473. https://doi.org/10.1021/jo961713w. [ Links ]
12. Armarego WL. Purification of Laboratory Chemicals. Butterworth-Heinemann; 2017. [ Links ]
13. De D, Krogstad FM, Byers LD, Krogstad DJ. Structure-Activity relationships for antiplasmodial activity among 7-substituted 4-aminoquinolines. J Med Chem. 1998;41(25):4918-4926. https://doi.org/10.1021/jm980146x. [ Links ]
14. de Souza MVN, Pais KC, Kaiser CR, Peralta MA, de L. Ferreira M, Lourenço MCS. Synthesis and in vitro antitubercular activity of a series of quinoline derivatives. Bioorg Med Chem. 2009 Feb;17(4):1474-1480. https://doi.org/10.1016/j.bmc.2009.01.013 [ Links ]
15. Kondaparla S, Soni A, Manhas A, Srivastava K, Puri SK, Katti S. Antimalarial activity of novel 4-aminoquinolines active against drug resistant strains. Bioorg Chem. 2017;70:74-85. https://doi.org/10.1016/j.bioorg.2016.11.010. [ Links ]
16. Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193(4254):673-675. https://doi.org/10.1126/science.781840. [ Links ]
17. Makler MT, Hinrichs DJ. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am J Trop Med Hyg. 1993;48(2):205-210. https://doi.org/10.4269/ajtmh.1993.48.205. [ Links ]
18. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1-2):55-63. https://doi.org/10.1016/0022-1759(83)90303-4. [ Links ]
19. Carter MD, Phelan VV, Sandlin RD, Bachmann BO, Wright DW. Lipophilic mediated assays for 3-hematin inhibitors. Comb Chem High Throughput Screen. 2010;13(3):285-292. https://doi.org/10.2174/138620710790980496. [ Links ]
20. Sandlin RD, Carter MD, Lee PJ, Auschwitz JM, Leed SE, Johnson JD, Wright DW. Use of the NP-40 detergent-mediated assay in discovery of inhibitors of 3-hematin crystallization. Antimicrob Agents Chemother. 2011;55(7):3363-3369. https://doi.org/10.1128/AAC.00121-11. [ Links ]
Received 14 September 2022
Revised 14 December 2022
Accepted 14 December 2022
* To whom correspondence should be addressed. Email: mags.blackie@ru.ac.za


{kind=link}