










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.76 Durban 2022
http://dx.doi.org/10.17159/0379-4350/2022/v76a12
RESEARCH ARTICLE
Molecular docking, dynamics, and quantum chemical study of vanillylacetone and beta-hydroxy ketone derivatives against Mpro of SARS-CoV-2
Saniyah AminI; Shabbir MuhammadII, *; Javed IqbalI, *; Sami UllahII; Abdullah G. Al-SehemiII; H AlgarniIII; Saleh S AlarfajiII; Khurshid AyubIV
IDepartment of Chemistry, University of Agriculture, Faisalabad, Pakistan
IIDepartment of Chemistry, College of Science, King Khalid University, Abha, Saudi Arabia
IIIDepartment of Physics, College of Science, King Khalid University, Abha, Saudi Arabia
IVDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, Pakistan
ABSTRACT
This study is carried out to find novel active drug candidates which can effectively bind to key residues of main protease (Mpro) of SARS-CoV-2. We performed molecular docking of fifty-seven (57) ligands from two classes: vanillylacetone and its derivatives and beta-hydroxy ketone derivatives against Mpro of SARS-CoV-2. We also docked three antiviral drugs as reference/benchmark drugs including remdesivir (RDV), chloroquine (CQ), and hydroxychloroquine (HCQ) against Mpro for comparison of inhibition tendencies of selected ligands. Binding energies of our reference drugs are as: CQ = -5.1 kcal mol-1 (with predicted inhibition constant (Ki pred) = 177 μmol), HCQ = -5.7 kcal mol-1 (Ki pred = 64.07 μmol) and RDV -6.3 kcal mol-1 (Ki pred = 13.95 μmol). We got remarkable results for our docked ligands as 79% of total ligands indicated binding energies better than CQ, 39 % better than both HCQ and CQ, and 19 % better than all reference drugs. More interestingly interaction analysis of eight best-docked ligands showed that they interacted with desired key residues of Mpro. We further selected the four best-docked ligands L1 = -6.6 kcal mol-1 (Ki pred =13.95 μmol), L6 = -7.0 kcal mol-1 (Ki pred = 7.08 μmol), L34 = -6.0 kcal mol-1 (Ki pred = 38.54 μmol), and L50 = -6.6 kcal mol-1 (Ki pred =13.95 umol) for further analysis by quantum chemical study, molecular dynamic (MD) simulations and ADMET analysis. We have also carried out MD-simulations of six more docked ligand L2, L14, L20, L36, L46 and L48 some of which were showing weak binding affinities and some average binding affinities to check their simulation behavior. Their RMSD, RMSF and binding free energy results were also quite satisfying. We believe the current investigation will evoke the scientific community and highlights the potential of selected compounds for potential use as antiviral compounds against Mpro of SARS-CoV-2.
Keywords: COVID-19; molecular docking; molecular dynamics; quantum analysis; SARS-CoV-2
INTRODUCTION
Pandemic is considered the worst case, in the domain of contagious diseases. The word pandemic originates from the Greek word pandemos where pan refers to everyone and demos means population. So, the term pandemos refers to the conception where everyone is susceptible to exposure of the disease, and a larger fraction of exposed people are likely to get sick. In December 2019, one such pandemic originated in Wuhan province of China which spread all around the world in no time and till present is uncontrolled.1 On 19 February 2021, 2 million deaths and 93 million new cases were globally reported.2 Human coronaviruses (HCoVs) were originally detected in 1965 in patients with associated symptoms of coined B814 and common flu. These HCoVs are enveloped, positive-sense single-stranded RNA, carrying 26-32 kilobases which is the largest sequence for any RNA virus and belongs to the family "Coronaviridae".3 During the previous two decades, coronaviruses have caused three massive eruptions including the SARS pandemic 2002-2003,4 the MERS pandemic 2012,5 and the ongoing SARS-CoV-2 outbreak.6 The genomic sequence of this injurious virus consists of a single strand of RNA and is quite comparable to other CoVs. It has been known that SARS-CoV-2 causes certain types of pneumonia which are also regarded as a huge danger to public health.
The 3-chymotrypsin like protease (3CLPro) or Mpro is the vital enzyme needed for viral proteolytic maturation, replication, and transcription. Mpro shows an important role in the breakdown of polyproteins. This cleavage generates functional proteins like exoribonuclease, endoribonuclease, and RNA polymerase.7 So, by targeting Mpro, viral maturation and multiplication will also stop and hence can be an effective drug target for novel drug discovery.8 Mpro enzyme is made up of a total of 306 amino acids consisting of three domains I, II, and III including a catalytic dyad of Cysteine145 and Histidine41.9 Structural analysis reveals that these amino acids and a few others located in the dimerization region and substrate-binding pocket of Mpro can be useful targets for the identification of active inhibitors in the treatment of COVID-19.10 Ginger (Zingiber officinale) is an influential plant having both nutritional and medicinal value which occurs naturally in many countries. Zingerone (vanillylacetone) and gingerol (beta-hydroxy ketone derivative) are essential constituents of Zingiber officinale and their pharmacological effects are above board i.e., they possess anticancer,11 anticoagulant,12 antiemetic,13 antioxidant,14 gastrointestinal,15 antitussive,16 antigenotoxic,17 cardiovascular,18 mutagenicity 19 and antimicrobial effects.16 The phenolic compounds in ginger, such as beta-hydroxy ketone and vanillylacetone, are primarily responsible for their health benefits. They have also been found effective against the treatment of neurodegenerative diseases,20 obesity,21 emesis, and chemotherapy-induced nausea.22 Beta-hydroxy ketone has also been found useful against some respiratory diseases.23 Furthermore, beta-hydroxy ketone and vanillylacetone have been used for the cure of anti-viral diseases.24 Because it takes a long time for novel antiviral medications to be licensed, numerous studies have been done to assess the efficacy of existing approved treatments against SARS-CoV-2.25 Several medicines have been discovered to have antiviral effects against SARS-CoV-2. Old antimalarial medications (hydroxychloroquine, chloroquine phosphate, and chloroquine) and viral RNA polymerase inhibitors are among them (favipiravir and remdesivir). So, considering their positive outcomes against COVID-19 patients we selected three drugs RDV, CQ, and HCQ as reference for comparison of the docking results of our ligands.
These days new effective inhibitors are being recognized by using advanced computational tools for the screening of large libraries of compounds.27 Experimental screening alone is not sufficient for the rapid discovery of efficient candidates and may not give better lead production.28 On the other hand, computational approaches play an important role in rapid drug discovery by employing some drug design i.e., molecular docking based on high throughput virtual screening is excessively used to explore lead compounds from large libraries of compounds.29 In this work, we have performed molecular docking of natural compounds, zingerone (vanillylacetone) and gingerol (beta-hydroxy ketone derivative) from Zingiber officinale and derivatives against Mpro regarding the medicinal value of these compounds. We have performed a quantum analysis of selected lead compounds by determining several chemical reactivities. Furthermore, we have also performed molecular dynamics of main protease and four selected ligand complexes for determining their stability and residual flexibility and assessed ADMET properties for getting non-toxic drug candidates. The detail of all steps involved in the whole process is given in Figure 1
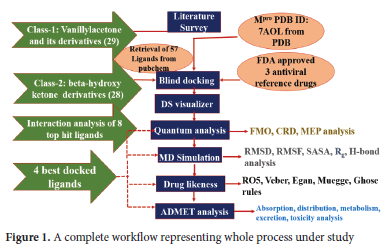
COMPUTATIONAL DETAILS
Selection of protein and ligands
The crystal structure of Mpro (PDB ID: 7AOL) was obtained from the protein data bank. This enzyme consists of 306 amino acids in the form of a single chain. This protein was further prepared in the Discovery Studio visualizer where inhibitor and water molecules were removed, and polar hydrogen was added to the protein structure. Ligands were downloaded in "SDF" format from PubChem and converted into pdbqt format via open babel before performing molecular docking in Autodock vina. Toxicity parameters were also evaluated from PubChem for the selection of ligands which are given in Table S2-S3 whereas information about all the ligands is presented in Table S4 and S5. Furthermore, ADMET properties of selected ligands were checked from Swissadme and pkCSM online server by using the smiles of those ligands retrieved from PubChem.
Molecular docking
Blind docking studies were done using the Autodock Vina program.30 Grid parameters were fixed with grid boundaries of 60 A x 60 A x 60 A unified at x-, y-, z-coordinates of -20, 25, and 35 A, respectively. These parameters were adjusted to cover the whole protein inside the box and the ligand can go to any available binding pocket. Different poses of each ligand and their respective binding energies (kcal mol-1) were calculated using Autodock Vina.30 All poses of ligands were visualized using BIOVIA Discovery Studio visualizer 31 however; the best pose with good binding affinity was used for performing interaction analysis of selected ligands. We replicated our docking results of selected eight ligands by using two other online docking servers i.e., Swissdock 32 and CB-dock.33 These replica results are given in Table S1 of supporting information. We got almost similar, reproducible results for all these eight ligands from Swissdock and Autodock vina. The predicted inhibition constants (Ki pred) were theoretically predicted from the binding energy (AG) using the formula:

where R is the universal gas constant (1.980 x 10-3 kcal mol-1 K-1) and T is the temperature (298.15 K).34
Quantum studies
A quantum chemical study for the selected ligands was performed in GAUSSIAN 09 35 for their structural analysis whereas molecular electrostatic-potential maps and frontier molecular orbitals were visualized via GaussView 06.36 The 3D structures of ligands were optimized to the lowest energy by using the density functional theory approach with B3LYP/6-311G* level of theory.
Molecular dynamics
Mpro and four selected ligands were further processed through molecular dynamics for comparing the stability of free form and its complex with selected ligands. Molecular dynamic (MD) simulations were performed on the most stable pose obtained from the docking studies using CHARMM force-field 37 in NAMD 38 while VMD 39 program was utilized for analyzing RMSD and RMSF. We used the water box of the TIP3P (three-site transferrable intermolecular potential) water model 40 to solvate the system and also performed neutralization by adding 4Na+ by autoionizing option in VMD before performing MD simulations. Stability and flexibility of protein and complexes were assessed from the values of their root mean square of deviation (RMSD), solvent accessible surface area (SASA), the radius of gyration (Rg), number of H-bonds, and root mean square of fluctuation (RMSF). Energy minimization was performed for 10 000 steps. Equilibration of each system was performed at temperature (310 K) and normal pressure (1 atm) for 1 ns. Afterward, a production run for 60 ns was conducted in constant temperature and volume. NAMD calculations were performed through a high-performance computing facility at King Abdullah University of Science & Technology (KAUST) in Thuwal, Saudi Arabia. MD results of selected complexes were also replicated thrice by using the same environment for performing MD simulations for all the replicas of every complex. RMSD and RMSF replicas of selected complexes are presented in supplementary material in Figure S1-S2.
RESULTS AND DISCUSSION
Work strategy based on the structure of Mpro
Mpro of SARS-CoV-2 is a dimer that consists of two monomers A (green) and B (blue) as shown in Figure 2 (a) and is active only in dimeric form. Both of the monomers are individually inactive, and dimerization is crucial for the functioning of Mpro.41 Each monomer consists of three domains I, II, and III where the asterisk represents domains of the B (blue) monomer in Figure 2 (a). Domain I covers residues 10-99 and domain II covers 100-182. A catalytic dyad in the form of His41 and Cys143 is also present in domain I and domain II. Catalytic residues are present in these domains I and II, so these are also known as catalytic domains. Domain III covers residues 198-303 and this domain III is connected via C-terminal to the catalytic domains. The dimerization is crucial for enzymatic activity. The retrieved Mpro structure is shown in Figure 2(a) along with labeled domains of both monomers. Structure of Mpro; a vital protein in SARS-CoV-2 functioning is highly preserved among different CoVs. There is a 96% similarity in the genomic sequence of Mpro of SARS-CoV and SARS-CoV-2.42
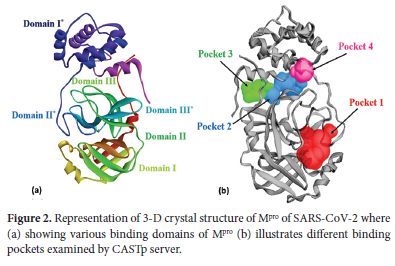
Among a total of 306 amino acid residues, only 12 residues differ in both. Interestingly, none of these 12 residues plays any significant role in the enzymatic activity of Mpro of SARS-CoV-2. Considering such a large similarity in genomic sequence it is expected that inhibitors effective against Mpro of SARS-CoV can be extremely useful in the inhibition of Mpro of SARS-CoV-2. Mpro of SARS-CoV also exists as a dimer and major residues with their associated roles are illustrated in Table 1 Any inhibitor targeting any of these residues will likely have a potential effect against inhibition of SARS-CoV-2. We performed blind docking of 57 ligands of two classes, class-1, 29 ligands from vanillylacetone and its derivatives, and class-2, 28 ligands from beta-hydroxy ketone derivatives as inhibitors of Mpro of SARS-CoV-2.

We also docked three reference antiviral drugs including RDV, CQ, and HCQ for comparing the inhibition potential of our docked ligands. We used the strategy of blind docking for finding some potential inhibitors which can block the catalytic activity of COVID-19 by blocking any of the following sites:
1) Substrate-binding pocket
2) Dimerization
3) Catalytic dyad
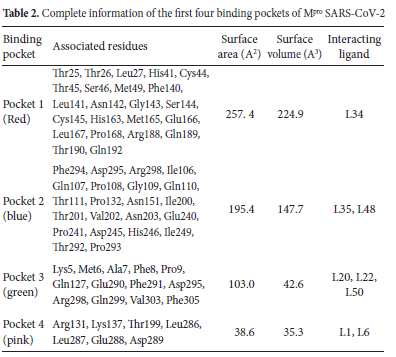
Mpro (PDB ID: 7AOL) consists of a total of 43 binding pockets, our analysis of docking results and interactions indicated that the top eight hit compounds showing the lowest binding energy (kcal mol-1) including L1, L6, L20, L22, L34, L35, L48, L50 interacted with first four binding and detail of these pockets is illustrated in Table 2 These pockets are colored differently in Figure 2(b) for better clarification of the location and relative sizes of these binding pockets. Further interaction analysis of these ligands has been presented above.
Interpretation of docking results
These classes vanillylacetone and beta-hydroxy ketone derivatives of Zingiber officinale were selected because of their known antiviral effects. The results of binding energies (kcal mol-1), inhibition constants (|imol) along PubChem ID of all docked ligands are shown in Table 3 The range of binding energies for our ligands was -4.4 to -7 kcal mol-1. RDV indicated binding energy of -6.3 kcal mol-1, HCQ as -5.7 kcal mol-1, and CQ as -5.1 kcal mol-1. Surprisingly, forty-five of our docked ligands showed binding affinities either higher or equal to CQ, twenty-two expressed higher than HCQ and CQ and eleven ligands were those which showed binding affinities either greater or equal to all three-reference drugs as shown in Table 3 It is also clear from Table 3our class 1 ligands showed binding affinities somewhat higher than class 2. These classes vanillylacetone and beta-hydroxy ketone derivatives of Zingiber officinale were selected because of their known antiviral effects. L6 from class 1 showed the highest binding affinity of -7 kcal mol-1 among all the docked ligands whereas L50 showed a maximum binding affinity of -6.6 kcal mol-1 among all class 2 ligands.
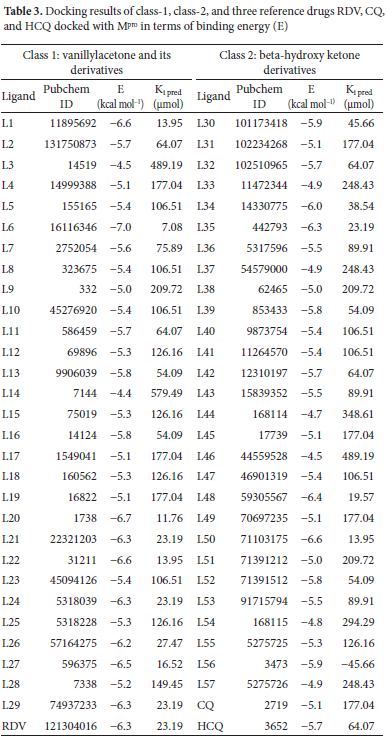
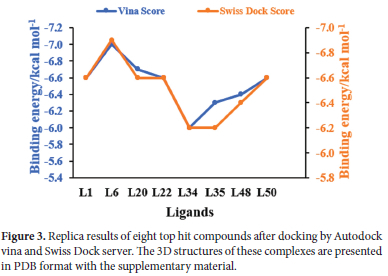
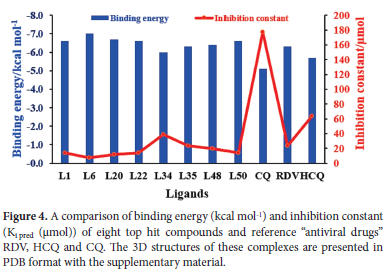
We further performed interaction analysis of eight ligands, four per class L1, L6, L20, L22 from class 1 and L34, L35, L48, L50 from class 2, which were showing higher binding affinities in their class. Their range of binding affinities in decreasing order is as: L6 > L20 > L1 = L22 = L50 > L48 > L35 > L34 whereas reverse will be the order for inhibition constant Ki (μmol). We replicated our docking results for these selected ligands by docking in three different docking tools i.e., auto dock vina, Swiss dock, and CB-Dock. These replica results are provided in Table S1 of supporting information. As shown in Figure 3 almost the same docking results were obtained by docking in Autodock Vina and Swiss dock. L6 is showing the highest binding affinity of -7 kcal mol-1 and the smallest Ki of 7.08 μmol whereas L34 is showing the lowest binding affinity of -6 kcal mol-1 and the highest Ki of 38.54 μmol among these eight ligands by docking in Autodock vina. As Ki denotes the amount of compound needed to inhibit at a half maximum level the smaller value of Ki shows that a smaller amount of drug will be sufficient for 50% inhibition and vice versa. Among our eight selected ligands, all are showing higher binding affinity (leading to stable complex formation) and lower Ki pred than all our reference antiviral drugs except L34 (which is showing the lower binding affinity and higher Ki pred than single reference RDV). We selected L34 for further study based on our interaction analysis. Figure 4shows a graphical comparison of the binding energies of our selected ligands and reference drugs.
Protein-Ligand Interaction analysis
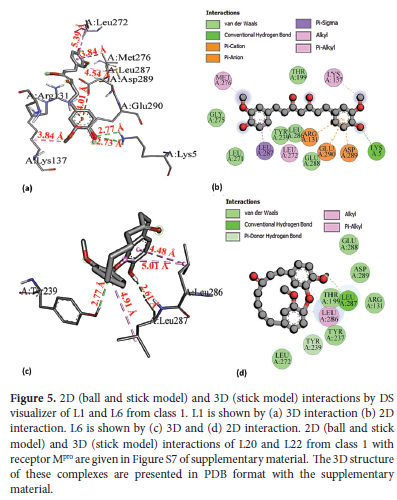
Interaction analysis of all selected ligands and reference RDV is given in Table 4 As RDV is showing the lowest binding energy among three reference drugs we performed a further comparison of docking results of our ligands with this reference. Four best-docked ligands including L1, L6 from class 1 and L34, L50 from class 2 were further filtered from these eight ligands for their detailed analysis. Total density representation figures of these eight ligands are presented in supplementary material in Figure S3-S6. The 2D and 3D interactions for the most stable first pose are shown in Figure 5(L1 and L6) and Figure 6(L34, L50) whereas interaction figures i.e., 3D and 2D and complete detail of interaction analysis for remaining ligands L20, L22, L35, and L48 are given in Figure S7 and S8 of supplementary material. PDB files of all selected ligand complexes are also given in zipped form in supporting material. L1 and L6 interacted with binding pocket 4 (pink) as illustrated in Table 2 L1 is stabilized by two hydrogen bonds with Lys5 (H donor) at 2.77 A and 2.73 A. L1 also forms four hydrophobic interactions: one hydrophobic bond of pi-sigma type with Leu287 (3.84 A), two hydrophobic-interactions of alkyl type; one with Met276 (4.54 A) and other with Lys137 (3.85 A) whereas one hydrophobic bond of n-alkyl nature is present with Leu272 (5.39 A). Leu287 (3.84 A) bond is expected to be strongest among all of these four hydrophobic interactions. L1 further displays three electrostatic interactions, two electrostatic interactions of n-anion type: one with Asp289 (4.01 A) and other with Glu290 (4.21 A), third electrostatic interaction is of n-cation nature with Arg131 (4.72 A). Asp289 (4.01 A) is expected to be the strongest among these three electrostatic interactions. Additionally, Van der Waals interactions are also present with some residues including i.e., Thr199, Leu286. L6 forms two H-bonds; one H-bond of conventional type is present with Leu287 (2.61 A) and other n-donor type H-bond is formed with Tyr239 (2.78 A). Leu287 (2.61 A) is expected to be stronger. L6 is also involved in three hydrophobic interactions. One alkyl-type hydrophobic interaction is found with Leu286 at 5.01 A while two n-alkyl type hydrophobic interactions are present with Leu286(4.49 A) and Leu287 at 4.91 A. Leu286 (4.49 A) is likely to be the strongest one among these three hydrophobic interactions.

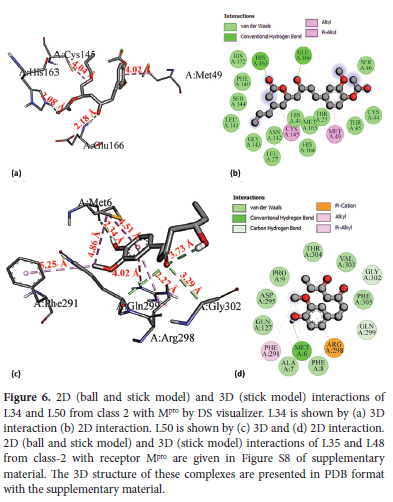
L34 interacts with binding pocket 1 (red) and makes two conventional H-bonds with His163 (2.08 A) and Glu166 (2.18 A). is the (2.08 A) being stronger one. L34 also forms two hydrophobic interactions: one alkyl-type hydrophobic interaction is formed with Cys145 (4.04 A) which is an active site residue and other hydrophobic interaction of n-alkyl type is formed with Met49 (4.02 A). Van der Waals interactions are also found with residues including His41, Thr25, Cys44, Ser46, and144, Leu141 an Asn142 etc. L50 forms four hydrogen bonds: one conventional type with Met6 (H-donor) at bond length of 2.34 A, two C-H types with Gln299 (H-donor) and Gly302 (H-donor) at 3.73 A and 3.29 A, respectively whereas one n-donor type H-bond is generated with Arg298 (H-donor) at 4.02 A. Met6 (2.34 A) is expected to be stronger. L50 also forms four hydrophobic interactions: one alkyl type with Met6 (4.86 A) and three n-alkyl type hydrophobic interactions are formed with Phe291 (5.25 A), Met6 (4.51 A) and Arg298 (4.92 A). Met6 (4.51 A) is likely to be the strongest among these hydrophobic interactions. Our reference antiviral RDV forms only four hydrogen bonds: three conventional type H-bonds are formed with Arg131 (H- donor), Tyr239(H-donor) and Leu287 (H-donor) at 2.54 A, 1.90 A and 2.68 A, respectively whereas fourth H-bond of C-H type is formed with Asn238 (H-acceptor) at 3.76 A.
It would be worthy to mention here that there are some common interacting residues in RDV, L1 and L6. Leu287 and Arg131 are common residues that are showing interaction in both RDV and L1 whereas Leu287 is common in both RDV and L6. Our all selected ligands are showing interactions with key residues mentioned in Table 1 These residues have fundamental roles in the functioning and maintenance of the structure of Mpro of SARS-CoV and are expected to be significant against inhibition of SARS-CoV-2. Chou et al. reported a full loss of dimerization and catalytic activity of Mpro of SARS-CoV due to mutation in Glu290.43 L1 is showing interaction with Glu290. Ding et al., reported that dimerization inhibitor of Mpro of SARS-CoV developed hydrophobic contacts with Met6 which played a significant contribution in the binding of inhibitor and loss of dimerization.50 Lin et al., reported that deletion of Arg298 and Gln299 significantly lowered the enzymatic activity to 1-2% of Mpro SARS-CoV.51 Shi et al., further confirmed that Arg298 plays a significant role in maintaining dimeric structure.45 Met6 and Arg298 are common residues showing interaction in L20, L22, L48, and L50 whereas Arg298 is also showing interaction in L35. L20, L22, and L50 are interacting with Gln299. Previous studies indicated that Glu166 acts as a linker between the substrate-binding pocket and dimeric interface and this connection might be universal in proteases of all CoVs.52 Tan et al., stated that the protonation state of His163 and His172 residing in substrate binding pocket, in turn, controls the conformation and size of substrate binding pocket.53 Hsu et al. further described that mutation in Cys145 affected dimerization and can also be fruitful in the inhibition of Mpro. Our L34 is showing interactions with these key residues Cys145, His163, and Glu166. Arg131, Leu286, and Leu287 are also known to have a role in dimerization, and mutation in these residues will have a detrimental effect on enzymatic activity.54 L1 is interacting with Leu287 and Arg131 while L6 shows interactions with Leu286 and Leu287. Based on our interaction analysis it can be justly stated that our ligands can be extremely useful against inhibition of SARS-CoV-2.
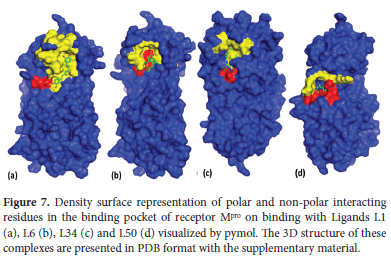
A quick view of the polar and nonpolar interacting residues of receptor Mpro on binding with our lead compounds is given in Figure 7 Polar interactions refer to H-bonds represented by red color and non-polar interactions include hydrophobic and other such interactions shown by yellow color. The blue color represents the entire protein surface. It can be seen in Figure 7maximum red area is shown in (d) where L50 is forming four H-bonds. Similarly, the yellow region is also by the number of non-polar interactions in each case. There is a significant yellow area in all figures and a maximum yellow area in (a) than in (d) just by their number of non-polar interactions.
Quantum analysis
DFT calculations were utilized for establishing optimized-structural parameters i.e., dihedral angle, bond length, and bond angle for potential lead compounds L1, L6, L34, L50 using B3LYP-6-311G* calculation. The two- and three-dimensional optimized molecular geometries of these lead compounds plotted in GaussView are presented in the supplementary material.
FMO and GCRD analysis of selected ligands L1, L6, L34, and L50
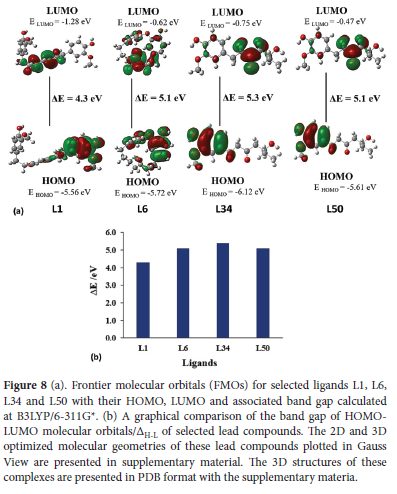
Molecular orbitals and their associated parameters i.e., chemical softness (S), hardness (N), electrophilicity index (w), etc are used for determining the reactivity trends.55 It also describes the electronic and optical properties of an organic compound 56. The energy of HOMO denotes the ability to donate electrons and the energy of LUMO indicates the tendency to accept electrons whereas the HOMO-LUMO band gap characterizes the chemical reactivity and kinetic stability of a molecule. In all these cases it can be seen that HOMO is located around electron-rich aryl group containing etheric or alcoholic moiety whereas LUMO is concentrated around the electron-poor unsaturated aliphatic chain or carbonyl groups. Some parameters i.e., electronegativity index (x), hardness (N), chemical potential etc as listed in Table S6 of supporting material depend on the gap of HOMO-LUMO 57 as stated by Koopmans theorem 58 Parr and Pearson interpretation 59 of DFT. Kinetic stability has a direct relationship with bandgap while chemical reactivity has an inverse relation. As shown in Figure 8our ligand L1 has the smallest HOMO-LUMO gap of 4.3 eV, L34 has the largest (5.3 eV) while L6 and L50 have a similar value (5.1 eV). Energy associated with HOMO indicates ionization potential (I) as HOMO donates electrons in any molecular interaction whereas the energy of LUMO represents electron affinity (A) as LUMO accepts electrons during a molecular interaction.60 As shown in Table S6 L1 has the smallest value of I as 5.56 and L34 has the highest, at 6.12 eV The range of I in decreasing order for these four selected ligands is as: L34 > L6 > L50 > L1. L1 possesses the highest value of A at 1.28 eV and L50 shows the smallest value of 0.4 eV. Order of A, for selected ligands in decreasing range, is L1 > L34 > L6 > L50 as shown in Table S6. Chemical potential (|) describes charge transfer in a molecular system that is present in the ground state.61 Chemical potential is directly related to chemical reactivity. L50 shows the highest μ-3.04 and L34 shows the lowest (-3.44 eV). Order of μ in decreasing range is as: L50 > L6 > L1 > L34. Electrophilicity index is based on thermodynamical properties of a molecule, it is a measure of the favorable variation in energy when a system gets saturated by adding electrons. In other words, it represents a corresponding decline in energy when electrons flow from HOMO (donor) towards LUMO (acceptor). L1 has the highest w of 2.73 and L50 has the lowest, at 1.81 eV. The order of w in decreasing range is as: L1 > L34 > L6 > L50. Electronegativity index represents the attraction of electrons in a covalent bond for any atom. Compounds having high x represent the high ratio of charge flow. L34 has the highest x value of 3.44 eV while L50 has the lowest (3.04 eV) and order in decreasing range can be represented as L34 > L1 > L6 > L50. Hardness depends on ionization energy (I) and electron affinity (A) of the molecular system. L1 shows the smallest n of 2.14 eV whereas L34 shows the largest (2.68 eV) and decreasing order of r for all these ligands is given as L34 > L50 = L6 > L1. The softness of reacting species depends on hardness. A compound having a smaller bandgap is considered softer and vice versa.62 L1 shows the maximum S of 0.23 and L34 shows the minimum 0.18 and order in decreasing range is as L1 > L6 = L50 > L34 as shown in Table S6.
Molecular electrostatic potential (MEP)
MEP is regarded as an excellent map representing molecular shape and size as well as regions of negative, positive, and neutral electrostatic potential via different color coding. Furthermore, MEP is considered an extremely useful descriptor in identifying sites for H-bonding as well as electrophilic and nucleophilic reactions.63 MEP is well suited for the analysis of enzyme-substrate and drug-receptor type of reactions which are initiated via recognition of one moiety by another. This recognition is in turn controlled by their MEPChemical potential.64 We calculated MEP for our selected ligands L1, L6, L34, and L50 at B3LYP, 6-311G* optimized structural geometry. MEP range in decreasing order is as; blue > green > orange > red. The red color indicates the region having the highest value of negative potential while blue shows the region of maximum positive potential. As shown in Figure 9red color is found at many sites in all selected ligands mainly around highly electronegative O which represents a favorable site for electrophilic attack. In the case of the L1 red region is around Asp289 and Glu290 where highly electronegative O atom of these amino acids is involved in electrostatic interaction with receptor Mpro. In the case of the L6 red region is primarily concentrated around Leu287. In the case of L34 His163, Glu166, and in the case of L50 Met6 which act as H-donor in the formation of H-bond with Mpro are present in the red region. The Blue region is dominantly present around H (in all cases) representing a suitable region for nucleophilic attack. The Blue region is mainly concentrated around H-bond donor Lys5 in the case of L1. In the case of L6, the blue region is present around H-donor Tyr239 involved in H-bond and Leu286 involved in hydrophobic interaction. Cys145 and Met49 are involved in hydrophobic interactions for L34, Arg298, and Met6 in the case of L50 which are involved in electrostatic and hydrophobic interactions with Mpro are also located in the blue region. The green color shows the area of zero potential.

Molecular Dynamics
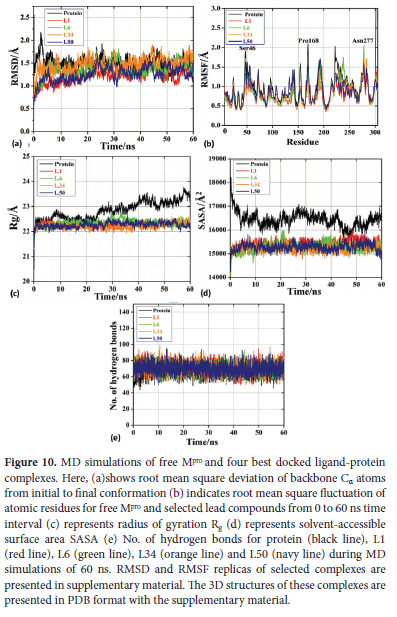
For investigating the stability and residual flexibility of free protein (apo form) and four selected ligand complexes L1, L6, L34, L50 their MD-simulations were performed for the period of 60 ns. Furthermore, we also performed MD-simulations of six more ligands L2, L14, L20, L36, L46 and L48. Of them L14, L46 are showing weakest binding affinity, two ligands L2, L36 are showing average and two L20, L48 are showing binding affinity closer to above 4 high affinity ligands complexes. We performed their MD-simulations to check how do they behave under similar conditions. Three molecular dynamics replicas were run for each ligand-protein complex in the same environment for obtaining reproducible MD results. MD replica of RMSD and RMSF for all these complexes are presented in supplementary material. Only a single reproducible RMSD, RMSF trajectory for all these complexes is given in Figure 10 Literature survey indicates that conclusions drawn from multiple shorter replicas are more consistent than those from a single longer simulation 65.
Calculation of RMSD and RMSF
RMSD acts as an indicator of stability of system whereas RMSF explains residual flexibility. For the RMSD analysis, we determined the deviation of backbone atoms of the protein during its initial to final conformation in free state as well as in complex with lead compounds. Stability of a system i.e., protein or ligand complex is inversely related to RMSD.66 MD simulations of apo protein shows an average RMSD of 0.5 A as shown in black line in Figure 10(a). This system shows maximum deviation of RMSD during first 5 ns where it deviates from 2.1 A at 3 ns to 1.3 A at 5 ns. After that it starts attaining stability and remains almost stable during whole remaining trajectory of 60 ns. Minimum RMSD of 1.1 A occurs at 51 ns. L1 complex shows an average RMSD of 0.44 A as shown by red line in Figure 10(a). It shows variation in RMSD during starting phase where RMSD keeps on rising from 0.76 A to 1.45 A from a period of 1 ns to 18 ns after that it gets stability and shows stable trajectory till 40 ns. During 18 ns to 40 ns RMSD oscillates between 1.45 to 1.02 A. At 43 ns system L1 complex reaches a maximum of 1.58 A and fluctuates between 1.58 to 1.02 A during last period of43-60 ns. L6 complex shows an average RMSD of 0.48 A as represented by green line in Figure 10(a). This complex is showing an increase in RMSD from a minimum of 0.8 A to a maximum of 1.81 A in the interval of 1-31 ns. At 32 ns it shows sudden decrease in RMSD to 1.25 A. L6 complex is showing RMSD in the range 1.2-1.7 A during the interval of 32-60 ns. Maximum RMSD of 2.24 A occurs at 24 ns. L34 complex shows an average RMSD of 0.45 A depicted by orange line in Figure 10(a). During first 10 ns system shows RMSD in the range 1.1-1.5 A. During the interval of 10-20 ns RMSD keeps rising and reaches to 1.84 A at 20 ns. It further decreases to 1.12 A at 31 ns and once again continue rising till it reaches to maximum of 1.89 A at 45 ns. This L34 system shows almost stable trajectory onward till 60 ns. L50 complex shows average RMSD of 0.44 A as shown by navy line in Figure 10(a). During first5 ns L50 shows an increase in RMSD from 0.8 to 1.4 A. This complex system shows most stable trajectory in the range 6-22 ns where it oscillates in the range 1-1.25 A. At 23 ns it reaches to maximum of 1.68 A and then decreases to 1 A at 30 ns. At 32 ns, it reaches to 1.45 A. During the interval of 32-43 ns, it oscillates in the range 1.1-1.55 A. Once again it shows deviation and reaches to 1.1 A at 45 ns. From onwards RMSD keeps fluctuating in the range 1.1-1.55 A. From above discussion and Figure 10(a) it is clear that all of our ligand complexes are showing RMSD smaller than 3 A which is an indication of their stable complexes 67. Ligand's RMSD (unbound form) has been given in Figure S1. In case of remaining six ligands L2, L14, L20, L36, L46 and L48 it is observed that maximum deviations are shown for L14 (wine line) in the region 33 ns to 55 ns from 2 to 2.7 A and L36 (pink line) in the region of 10 to 20 ns from 1 to 1.7 A , while remaining ligands do not show much deviation in RMSD as shown in Figure 11(a). Literature survey indicated that CQ showed good affinity towards Mpro during MD simulation. Its MD trajectories remain quite stable at the initial phase of MD and deviates around 150 ns.68 Literature survey also indicates that HCQ-Mpro shows average RMSD of 3.5 A during 100 ns of MD-simulation.26 Previous studies have shown that for RMSD of RDV was determined to be in the range of 2.4 to 5.4 A during the 30 ns of MD simulation.69
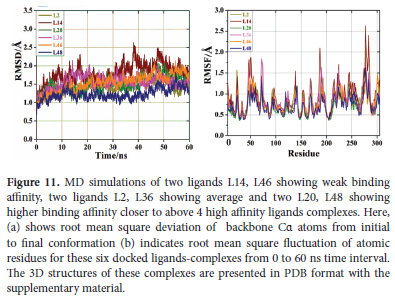
RMSF indicates amino acids fluctuation in overall system as shown in Figure 10(b). The higher the value of RMSF, the lesser is the stability (higher flexibility) of the system and vice versa.70 Only few significant deviations were observed for amino acid residues with respect to backbone Ca atoms during whole simulation trajectory. Almost all our ligand-protein complexes are showing stable RMSF trajectory with no significant residual fluctuation as shown in Figure 10(b). In case of free protein (black line) three fluctuation peaks are observed for Ser46, Pro168 and Asn221 at 1.9, 2.08 and 2 A, respectively. L1 complex (red line in b) also shows three fluctuation peaks for Ser46 at 1.85 A, Pro168 and Asn221 at 2.85 A. L6 complex (green line in b) shows four fluctuation peaks at 1.7, 1.6, 1.7 and 2.05 A for Ser46, Ala193, Asn238 and Asn277. L34 complex (orange line in b) shows two fluctuation peaks for Asn277 and Gly283 at 1.8 and 1.7 A. In case of L50 complex (navy line) only one fluctuation peak is observed for Gln192 at 1.7 A. RMSF analysis also demonstrates that our ligands are forming quite stable complexes. In case of free protein (black line) three fluctuation peaks are observed for Ser46, Pro168 and Asn221 at 1.9, 2.08 and 2 A, respectively. L1 complex (red line in b) also shows three fluctuation peaks for Ser46 at 1.85 A, Pro168 and Asn221 at 2.85 A. L6 complex (green line in b) shows four fluctuation peaks at 1.7, 1.6, 1.7 and 2.05 A for Ser46, Ala193, Asn238 and Asn277. L34 complex (orange line in b) shows two fluctuation peaks for Asn277 and Gly283 at 1.8 and 1.7 A. In case of L50 complex (navy line) only one fluctuation peak is observed for Gln192 at 1.7 A. For the remaining six ligands L2, L14, L20, L36, L46 and L48 it is observed that just like RMSD maximum fluctuations in RMSF of 2.7 A are shown for L14 (wine line) in Asn277 and for L36 (pink) maximum RMSF of 1.7 A is shown at Asn77. While remaining ligands do not show any significant fluctuations in RMSF as shown in Figure 11 (b). In case of RDV atoms in the range of 10 to 18 (RMSF>1.5) and atoms in the range of 35-42 (RMSF>1.0) are significantly fluctuating.69 RMSF analysis also demonstrates that our ligands are forming quite stable complexes.
Calculation of Rg, SASA and No. of H-bonds
For the prediction of compactness of protein-ligand complexes, SASA and Rg were also calculated. SASA predicts the solvent accessibility or unfolding of protein during MD simulations. In other words, SASA gives an idea about extent of solvent-protein interaction. Radius of gyration is a vital parameter for the evaluation of compactness and structural integrity of studied systems. Radius of gyration is characterized as mass weighted root-mean square distance of set of atoms from mutual center-of-mass.71 As shown in Figure 10(c) and (d) binding of ligands with the protein leads to a tremendous decrease in Rg and SASA values in comparison to the free protein. This factor also supports the compactness or stability of our complexes. Stability of these complexes is further confirmed by the evaluation of the number of H-bonds during MD-simulations. As shown in Figure 10(e) our complexes showed greater number of H-bonds in the range 50-95 than free protein, which showed H-bonds in the range 45-85. This parameter is again in favor of the stability of complexes.
Calculation of binding Free energy by MMPBSA method
Previous studies have shown that reference CQ showed binding free energy of -8.08 kcal mol-1.72 Free energy of association of selected Mpro complexes was evaluated by MM-PBSA method. The CaFE 1(calculation of free energy) plugin 73 in VMD was used to calculate the MMPBSA binding free energies from MD trajectories where sampling of 100 frames/snapshots were taken over a period of 60 ns simulations. Absolute binding-free energies are assessed by:

This method links solvation models, calculations of non-polar-solvation free-energy and explicit Poisson-Boltzmann analysis to assess the free energy of binding.74

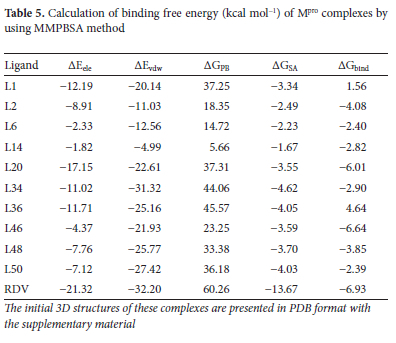
where EMM and GPBSA represent average molecular-mechanical energy and solvation-free-energy, respectively. Further details are very common and provided in supporting information for the purpose of brevity. Table 5shows that L46 is the most stable complex and L36 is the least. L2, L6, L14, L20, L34, L46, L48 and L50 show negative binding free energies whereas L1, L36 show positive. "Order of stability of complexes based on the values of binding free energy is: RDV > L46 > L20 > L2 > L48 > L34 > L14 > L6 > L50 > L1> L36. Their order of stability compared to the docking binding affinities from the Table 3is completely different, as expected." As discussed above L34 is interacting with key residues of Mpro i.e. His41, His163, Cys145, Glu166 and results of all the MD parameters also favor its quite stable complex formation. This stability is further proved by Figure 11.
Conformational changes in binding pocket of Mpro on binding with L34 during various time intervals of MD simulations from 0 to 60 ns are presented in supplementary material. These non-significant changes in binding pocket indicate that L34 is forming quite stable complex with Mpro and hence can act as an active inhibitor of Mpro. Our other selected ligands are also interacting with key residues and showing stable MD simulations. Cell boundary parameters are given in supporting information. Moreover, solvated, and ionized forms of these lead compounds and Mpro during MD simulations are presented in supplementary material.
Drug-likeness based on physicochemical properties
We evaluated drug likeness of our selected compounds and reference drugs via five different drug-likeness rules, each of which has its own conditions. Only L34 shows one Lipinski and one Veber violation (two violations of these parameters are acceptable so one violation of L34 does not count much 75). However, our reference antiviral drug RDV is showing two Lipinski, Veber and Ghose, three Muegge and one Egan violation. This prediction of physicochemical properties strongly favors that our selected ligands have full potential to act as drug candidates. Detail of these rules and results of different parameters for our lead compounds is presented in supplementary material (Table S7).
ADMET study
For efficient absorption of a drug candidate from gastrointestinal tract into systemic circulation (SC) it must satisfy several conditions.
Absorption
Some factors controlling the absorption of drug are gastrointestinal (GI) absorption, Log S, TPSA and Log P. For a compound to show appropriate absorption all these factors must be in optimal range. Our selected compounds are showing good GI absorption (in comparison to reference molecules) in the range 76-96%, Log S which shows water solubility is also in optimal range (-2 to -4.6) less than -5, Log P should be less than 5, the range of this factor for our selected lead compounds is (1.4-4.4) while TPSA should be less than 140 A2, our selected ligands have this value (55.8-96.2) as shown in Figure S13. So, all our selected ligands are complying with these factors which is a sign of their good drugability. These factors are also clear from bioavailability radar given in Figure S14-15.
Distribution
Some important factors regulating distribution of drug in the body include P-gp (permeability glycoprotein) and BBB permeation (blood-brain barrier permeation). All our selected candidates are acting as non-substrate to P-gp except L6 as shown in Table S8. L1 and L34 are impermeant to brain while L6 and L50 are BBB permeant as illustrated in Table S8. Both these factors are well illustrated by boiled egg in Figure S13.
Metabolism
Metabolism has key role in effective supply of drug molecules to the target sites which is accomplished by several enzymes most importantly by CYP450 76. Inhibition of these enzymes by any drug results in disturbance of metabolism of that drug and in severe cases even arrhythmia occurs.77 Three of these enzyme interactions were assessed for our selected ligands and reference drugs as shown in Table S8. All selected ligands are non-inhibitor of CYP3D4, CYP2C19 and CYP1A2 except L34 which causes inhibition of CYP2C19 and L6 which shows inhibition to CYP1A2.
Excretion
Two important factors indicating excretion of drug are TC (total clearance) and renal OCT2 (organic cation transporter 2). Our selected ligands are showing TC in the range 0.1-0.6 and are comparable to the value of this parameter for reference molecules given in Table S8. It can be seen in Table S8 except L34 all our selected ligands are non-substrate to OCT2.
Toxicity
This is a crucial factor for the selection of lead compound. A drug candidate is safe if it does not cause any mutagenic effect (AMES toxicity), skin effect (skin sensitization), liver damage (hepatotoxicity) and most importantly any side effect to heart (hERG I and II inhibition). As illustrated in Table S8 our selected ligands are fulfilling all these criteria of non-toxicity, showing even superior properties than reference drug molecules.
CONCLUSION
In summary, we investigated fifty-seven (57) ligands (vanillylacetone and beta hydroxy ketone derivatives) against Mpro of SARS-CoV-2 using together molecular docking, molecular dynamics methods and quantum chemical approaches for predicting active drug candidates which can affectively bind to the key residues of Mpro. Additionally, we analyzed the efficiency of three reference drugs having antiviral potential which have proven helpful against COVID-19 patients, for comparing inhibition potential of our docked ligands. The docking results i.e., binding energy and inhibition constant of our reference drugs were as: RDV = -6.3 kcal mol-1 (Ki pred = 23.19 μmol), HCQ = -5.7 kcal mol-1 (Ki pred = 64.07 μmol) and CQ = -5.1 kcal mol-1 (Ki pred = 177 |mol). Interestingly, molecular docking results illustrated that among all docked docked ligands and reference compounds, about 79% of ligands indicated binding energies better than CQ, 39% showed better than both HCQ and CQ, and 19% indicated better than all reference drugs. We further selected eight (8) ligands (four per class) which were showing the best binding affinities in their respective class and performed their interaction analysis. Remarkably, all of them interacted with the key residues of Mpro known to play key role in inhibition of Mpro. It was noticed that unlike reference RDV which formed only hydrogen bonds with Mpro our all ligands formed many other interactions i.e., hydrophobic, electrostatic, and Van der Waals etc. We further performed detailed analysis of four selected ligands (best docked) L1 = -7 kcal mol-1 (Ki pred = 7.08 |imol), L6 = -6.6 kcal mol-1 (Ki pred = 13.95 |imol), L34 = -6 kcal mol-1 (Ki pred = 38.54 μmol), L50 = -6.6 kcal mol-1 (Ki pred = 13.95 μmol). We have also studied the molecular structures of selected ligands via quantum chemical study. Quantum chemical study based on FMOs, GCRD and MEP analysis indicated that the selected ligands are quite reactive and kinetically enough stable. The MD simulations, assessment of drug likeness and ADMET profile were also done for best docked ligands. Analysis of MD simulations based on RMSD, RMSF, Rg, SASA and H-bonds plot showed that all ligand complexes are stable and do not show any significant residual fluctuations. Assessment of drug likeness by utilizing all five-important drug likeness prediction rules proved that our selected ligands fulfil all criteria of draggability. We have also carried out MD-simulations of six more docked ligand L2, L14, L20, L36, L46 and L48 some of which were showing weak binding affinities and some average binding affinities to check their simulation behavior.
Their RMSD, RMSF and binding free energy results were also quite satisfying. Based on detailed analysis of all studied parameters, we can fairly say that our studied ligands have great potential against inhibition of SARS-CoV-2.
ACKNOWLEDGEMENT
The authors extend their appreciation to the Institute of Research and Consulting Studies at King Khalid University for supporting this research through grant number 2-N-20/22 and the support of Research Center for Advanced Materials Science is highly acknowledged. For computer time, this research used the resources of the Supercomputing Laboratory at King Abdullah University of Science & Technology (KAUST) in Thuwal, Saudi Arabia.
SUPPLEMENTARY MATERIAL
Supplementary information for this article is provided in the online supplement.
REFERENCES
1. Loeffelholz MJ, Tang Y-W. Laboratory diagnosis of emerging human coronavirus infections-the state of the art. Emerg Microbes Infect. 2020; 9(1):747-756. https://doi.org/10.1080/22221751.2020.1745095 [ Links ]
2. C. COVID. R. Team, Allergic reactions including anaphylaxis after receipt of the first dose of Moderna COVID-19 Vaccine-United States, December 21, 2020-January 10, 2021. MMWR Morb Mortal Wkly Rep. 2021;70(4):125-129. https://doi.org/10.15585/mmwr.mm7004e1 [ Links ]
3. Li F. Receptor recognition and cross-species infections of SARS coronavirus. Antiviral Res. 2013;100(1):246-254. https://doi.org/10.1016/j.antiviral.2013.08.014 [ Links ]
4. Drosten C, Günther S, Preiser W, Van Der Werf S, Brodt H-R, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967-1976. https://doi.org/10.1056/NEJMoa030747 [ Links ]
5. Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814-1820. https://doi.org/10.1056/NEJMoa1211721 [ Links ]
6. Chan JF-W, Kok K-H, Zhu Z, Chu H, To KK-W, Yuan S, Yuen K-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221-236. https://doi.org/10.1080/22221751.2020.1719902 [ Links ]
7. Khan P, Parkash A, Islam A, Ahmad F, Hassan MI. Molecular basis of the structural stability of hemochromatosis factor E: A combined molecular dynamic simulation and GdmCl-induced denaturation study. Biopolymers. 2016;105(3):133-142. https://doi.org/10.1002/bip.22760 [ Links ]
8. Zhang Y-Z, Holmes EC. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. 2020;181(2):223-227. https://doi.org/10.1016/j.cell.2020.03.035 [ Links ]
9. Dai W, Zhang B, Jiang X-M, Su H, Li J, Zhao Y, Xie X, Jin Z, Peng J, Liu F, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368(6497):1331-1335. https://doi.org/10.1126/science.abb4489 [ Links ]
10. Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science. 2020;368(6489):409-412. https://doi.org/10.1126/science.abb3405 [ Links ]
11. Miyoshi N, Nakamura Y, Ueda Y, Abe M, Ozawa Y, Uchida K, Osawa T. Dietary ginger constituents, galanals A and B, are potent apoptosis inducers in Human T lymphoma Jurkat cells. Cancer Lett. 2003;199(2):113-119. https://doi.org/10.1016/S0304-3835(03)00381-1 [ Links ]
12. Surh Y-J, Park K-K, Chun K-S, Lee L, Lee E, Lee SS. Anti-tumor-promoting activities of selected pungent phenolic substances present in ginger. J Environ Pathol Toxicol Oncol. 1999;18:131-139. [ Links ]
13. Janssen P, Meyboom S, Van Staveren W, De Vegt F, Katan M. Consumption of ginger (Zingiber officinale roscoe) does not affect ex vivo platelet thromboxane production in humans. Eur J Clin Nutr. 1996;50:772-774. [ Links ]
14. Micklefield G, Redeker Y, Meister V, Jung O, Greving I, May B. Effects of ginger on gastroduodenal motility. Int J Clin Pharmacol Ther. 1999;37:341-346. [ Links ]
15. Fuhrman B, Rosenblat M, Hayek T, Coleman R, Aviram M. Ginger extract consumption reduces plasma cholesterol, inhibits LDL oxidation and attenuates development of atherosclerosis in atherosclerotic, apolipoprotein E-deficient mice. J Nutr. 2000;130(5):1124-1131. https://doi.org/10.1093/jn/130.5.1124 [ Links ]
16. Funk JL, Frye JB, Oyarzo JN, Timmermann BN. Comparative effects of two gingerol-containing Zingiber officinale extracts on experimental rheumatoid arthritis. J Nat Prod. 2009;72(3):403-407. https://doi.org/10.1021/np8006183 [ Links ]
17. Kim J-K, Kim Y, Na K-M, Surh Y-J, Kim T-Y. [6]-Gingerol prevents UVB-induced ROS production and COX-2 expression in vitro and in vivo. Free Radic Res. 2007;41(5):603-614. https://doi.org/10.1080/10715760701209896 [ Links ]
18. Srivastava K, Mustafa T. Ginger (Zingiber officinale) and rheumatic disorders. Med Hypotheses. 1989;29(1):25-28. https://doi.org/10.1016/0306-9877(89)90162-X [ Links ]
19. Beg T, Siddique YH, Ara G, Gupta J, Afzal M. Antigenotoxic effect of genistein and gingerol on genotoxicity induced by norethandrolone and oxandrolone in cultured human lymphocytes. Int J Pharmacol. 2008;4(3):177-183. https://doi.org/10.3923/ijp.2008.177.183 [ Links ]
20. Ho S-C, Chang K-S, Lin C-C. Anti-neuroinflammatory capacity of fresh ginger is attributed mainly to 10-gingerol. Food Chem. 2013;141(3):3183-3191. https://doi.org/10.1016/j.foodchem.2013.06.010 [ Links ]
21. Suk S, Kwon GT, Lee E, Jang WJ, Yang H, Kim JH, Thimmegowda N, Chung MY, Kwon JY, Yang S, et al. a polyphenol present in ginger, suppresses obesity and adipose tissue inflammation in high-fat diet-fed mice. Mol Nutr Food Res. 2017;61(10):1700139. https://doi.org/10.1002/mnfr.201700139 [ Links ]
22. Walstab J, Krüger D, Stark T, Hofmann T, Demir I, Ceyhan G, Feistel B, Schemann M, Niesler B. Ginger and its pungent constituents non-competitively inhibit activation of human recombinant and native 5-HT3 receptors of enteric neurons. Neurogastroenterol Motil. 2013;25(5):439-447. https://doi.org/10.1111/nmo.12107 [ Links ]
23. Townsend EA, Siviski ME, Zhang Y, Xu C, Hoonjan B, Emala CW. Effects of ginger and its constituents on airway smooth muscle relaxation and calcium regulation. Am J Respir Cell Mol Biol. 2013;48(2):157-163. https://doi.org/10.1165/rcmb.2012-0231OC [ Links ]
24. Ahkam AH, Hermanto FE, Alamsyah A, Aliyyah IH, Fatchiyah F. Virtual prediction of antiviral potential of ginger (Zingiber officinale) bioactive compounds against spike and MPro of SARS-CoV2 protein. Berkala Penelitian Hayati. 2020;25(2):52-57. https://doi.org/10.23869/bphjbr.25.2.20207 [ Links ]
25. Eweas AF, Alhossary AA, Abdel-Moneim AS. Molecular docking reveals Ivermectin and Remdesivir as potential repurposed drugs against SARS-CoV-2. Front Microbiol. 2021;11:592908. https://doi.org/10.3389/fmicb.2020.592908 [ Links ]
26. Mishra D, Maurya RR, Kumar K, Munjal NS, Bahadur V, Sharma S, Singh P, Bahadur I. Structurally modified compounds of hydroxychloroquine, remdesivir and tetrahydrocannabinol against main protease of SARS-CoV-2, a possible hope for COVID-19: docking and molecular dynamics simulation studies. J Mol Liq. 2021;335:116185. https://doi.org/10.1016/j.molliq.2021.116185 [ Links ]
27. Shamsi A, Mohammad T, Anwar S, AlAjmi MF, Hussain A, Rehman M, Islam A, Hassan M. Glecaprevir and Maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: possible implication in COVID-19 therapy. Biosci Rep. 2020;BSR20201256. https://doi.org/10.1042/BSR20201256 [ Links ]
28. Padhi AK, Tripathi T. Can SARS-CoV-2 accumulate mutations in the S-Protein to increase pathogenicity? ACS Pharmacol Transl Sci. 2020;3(5):1023-1026. https://doi.org/10.1021/acsptsci.0c00113 [ Links ]
29. Mohammad T, Arif K, Alajmi MF, Hussain A, Islam A, Rehman MT, Hassan I. Identification of high-affinity inhibitors of pyruvate dehydrogenase kinase-3: towards therapeutic management of cancer. J Biomol Struct Dyn. 2021;39(2):586-594. https://doi.org/10.1080/07391102.2020.1711810 [ Links ]
30. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455-461. [ Links ]
31. Lalit S, Vyomesh J. Comparative docking analysis of rational drugs against COVID-19 main protease. [Preprint]. ChemRxiv. 2020. https://doi.org/10.26434/chemrxiv.12136002.v1 [ Links ]
32. Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39 suppl:W270-W277. https://doi.org/10.1093/nar/gkr366 [ Links ]
33. Liu Y, Grimm M, Dai W, Hou M, Xiao Z-X, Cao Y. CB-Dock: a web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol Sin. 2020;41(1):138-144. https://doi.org/10.1038/s41401-019-0228-6 [ Links ]
34. Ortiz CLD, Completo GC, Nacario RC, Nellas RB. Potential inhibitors of galactofuranosyltransferase 2 (GlfT2): molecular docking, 3D-QSAR, and in silico ADMETox Studies. Sci Rep. 2019;9(1):17096. https://doi.org/10.1038/s41598-019-52764-8 [ Links ]
35. Frisch MJEA, Trucks GW, Schlegel HB, Scuseria GB, Robb MA, Cheeseman JR, Scalmani G. Gaussian 09, revision D. 01. Wallingford CT: Gaussian Inc. 2009. [ Links ]
36. Dennington R, Keith T , Millam J. GaussView, version. Shawnee Mission KS: Semichem Inc. 2009. [ Links ]
37. Vanommeslaeghe K, MacKerell AD Jr, Automation of the CHARMM General Force Field. (CGenFF) I: bond perception and atom typing. J Chem Inf Model. 2012;52(12):3144-3154. https://doi.org/10.1021/ci300363c. [ Links ]
38. Phillips JC, Hardy DJ, Maia JD, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Hénin J, Jiang W, et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys. 2020;153(4):044130. https://doi.org/10.1063/5.0014475 [ Links ]
39. Humphrey W, Dalke A, Schulten KDA, Schulten K. VMD-visual molecular dynamics. J Mol Graph. 1996;14(1):33-38. https://doi.org/10.1016/0263-7855(96)00018-5 [ Links ]
40. Vassetti D, Pagliai M, Procacci P. Assessment of GAFF2 and OPLS-AA general force fields in combination with the water models TIP3P, SPCE, and OPC3 for the solvation free energy of druglike organic molecules. J Chem Theory Comput. 2019;15(3):1983c1995. https://doi.org/10.1021/acs.jctc.8b01039 [ Links ]
41. Goyal B, Goyal D. Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb Sci. 2020;22(6):297-305. https://doi.org/10.1021/acscombsci.0c00058 [ Links ]
42. Goyal B, Goyal D. Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb Sci. 2020;15(6):297-305. https://doi.org/10.1021/acscombsci.0c00058 [ Links ]
43. Chou C-Y, Chang H-C, Hsu W-C, Lin T-Z, Lin C-H, Chang G-G. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43(47):14958-14970. https://doi.org/10.1021/bi0490237 [ Links ]
44. Chen S, Hu T, Zhang J, Chen J, Chen K, Ding J, Jiang H, Shen X. Mutation of Gly-11 on the dimer interface results in the complete crystallographic dimer dissociation of severe acute respiratory syndrome coronavirus 3C-like protease: crystal structure with molecular dynamics simulations. J Biol Chem. 2008;283(1):554-564. https://doi.org/10.1074/jbc.M705240200 [ Links ]
45. Shi J, Sivaraman J, Song J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J Virol. 2008;82(9):4620-4629. https://doi.org/10.1128/JVI.02680-07 [ Links ]
46. Barrila J, Gabelli SB, Bacha U, Amzel LM, Freire E. Mutation of Asn28 disrupts the dimerization and enzymatic activity of SARS 3CLpro. Biochemistry. 2010;49(20):4308-4317. https://doi.org/10.1021/bi1002585 [ Links ]
47. Huang C, Wei P, Fan K, Liu Y, Lai L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43(15):4568-4574. https://doi.org/10.1021/bi036022q [ Links ]
48. Shan Y-F, Li S-F, Xu G-J. A novel auto-cleavage assay for studying mutational effects on the active site of severe acute respiratory syndrome coronavirus 3C-like protease. Biochem Biophys Res Commun. 2004;324(2):579-583. https://doi.org/10.1016/j.bbrc.2004.09.088 [ Links ]
49. Hsu M-F, Kuo C-J, Chang K-T, Chang H-C, Chou C-C, Ko T-P, Shr H-L, Chang G-G, Wang AH-J, Liang P-H. Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem. 2005;280(35):31257-31266. https://doi.org/10.1074/jbc.M502577200 [ Links ]
50. Ding L, Zhang X-X, Wei P, Fan K, Lai L. The interaction between severe acute respiratory syndrome coronavirus 3C-like proteinase and a dimeric inhibitor by capillary electrophoresis. Anal Biochem. 2005;343(1):159-165. https://doi.org/10.1016/j.ab.2005.04.027 [ Links ]
51. Lin P-Y, Chou C-Y, Chang H-C, Hsu W-C, Chang G-G. Correlation between dissociation and catalysis of SARS-CoV main protease. Arch Biochem Biophys. 2008;472(1):34-42. https://doi.org/10.1016/j.abb.2008.01.023 [ Links ]
52. Cheng S-C, Chang G-G, Chou C-Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys J. 2010;98(7):1327-1336. https://doi.org/10.1016/j.bpj.2009.12.4272 [ Links ]
53. Tan J, Verschueren KH, Anand K, Shen J, Yang M, Xu Y, Rao Z, Bigalke J, Heisen B, Mesters JR, et al. pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J Mol Biol. 2005;354(1):25-40. https://doi.org/10.1016/j.jmb.2005.09.012 [ Links ]
54. Bhat ZA, Chitara D, Iqbal J, Sanjeev B, Madhumalar A. Targeting allosteric pockets of SARS-CoV-2 main protease Mpro. J Biomol Struct Dyn. 2021;40(14):6603-6618. https://doi.org/10.1080/07391102.2021.1891141 [ Links ]
55. Choudhary N, Bee S, Gupta A, Tandon P. Comparative vibrational spectroscopic studies, HOMO-LUMO and NBO analysis of N-(phenyl)-2, 2-dichloroacetamide, N-(2-chloro phenyl)-2, 2-dichloroacetamide and N-(4-chloro phenyl)-2, 2-dichloroacetamide based on density functional theory. Comput Theor Chem. 2013;1016:8-21. https://doi.org/10.1016/j.comptc.2013.04.008 [ Links ]
56. Padmaja L, Ravikumar C, Sajan D, Hubert Joe I, Jayakumar V, Pettit G, Faurskov Nielsen O. Density functional study on the structural conformations and intramolecular charge transfer from the vibrational spectra of the anticancer drug combretastatin-A2. J Raman Spectrosc. 2009;40(4):419-428. https://doi.org/10.1002/jrs.2145 [ Links ]
57. Parr RG, Zhou Z. Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters. Acc Chem Res. 1993;26(5):256-258. https://doi.org/10.1021/ar00029a005 [ Links ]
58. Pearson RG. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci USA. 1986;83(22):8440-8441. https://doi.org/10.1073/pnas.83.22.8440 [ Links ]
59. Pearson RG. The HSAB principle-more quantitative aspects. Inorg Chim Acta. 1995;240(1-2):93-98. https://doi.org/10.1016/0020-1693(95)04648-8 [ Links ]
60. Fukui K, Yonezawa T, Shingu H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J Chem Phys. 1952;20(4):722-725. https://doi.org/10.1063/1.1700523 [ Links ]
61. Parr RG, Donnelly RA, Levy M, Palke WE. Electronegativity: the density functional viewpoint. J Chem Phys. 1978;68(8):3801-3807. https://doi.org/10.1063/1.436185 [ Links ]
62. Fleming I. Frontier orbitals and organic chemical reactions. London: Wiley. 1977. [ Links ]
63.Scrocco E, Tomasi J. Electronic molecular structure, reactivity and intermolecular forces: An euristic interpretation by means of electrostatic molecular potentials. In: Löwdin P-O, editor. Advances in Quantum Chemistry. Vol. 11. Academic Press; 1978. p. 115-193. https://doi.org/10.1016/S0065-3276(08)60236-1 [ Links ]
64. Scrocco E, Tomasi J. New Concepts II. Topics in Current Chemistry Fortschritte der Chemischen Forschung. Vol. 42. Berlin, Heidelberg: Springer; 1973. The electrostatic molecular potential as a tool for the interpretation of molecular properties. https://doi.org/10.1007/3-540-06399-4_6 [ Links ]
65. Knapp B, Ospina L, Deane CM. Avoiding false positive conclusions in molecular simulation: the importance of replicas. J Chem Theory Comput. 2018;14(12):6127-6138. https://doi.org/10.1021/acs.jctc.8b00391 [ Links ]
66. Aier I, Varadwaj PK, Raj U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci Rep. 2016;6(1):34984. https://doi.org/10.1038/srep34984 [ Links ]
67. Kufareva I, Abagyan R. Methods of protein structure comparison. In: Orry A, Abagyan R, editors. Homology Modeling. Methods in Molecular Biology. Vol. 857. Humana Press; 2011. https://doi.org/10.1007/978-1-61779-588-6_10 [ Links ]
68. Luo S, Huang K, Zhao X, Cong Y, Zhang JZ, Duan L. Inhibition mechanism and hot-spot prediction of nine potential drugs for SARS-CoV-2 M pro by large-scale molecular dynamic simulations combined with accurate binding free energy calculations. Nanoscale. 2021;13(17):8313-8332. https://doi.org/10.1039/D0NR07833F [ Links ]
69. Rai H, Barik A, Singh YP, Suresh A, Singh L, Singh G, Nayak UY, Dubey VK, Modi G. Molecular docking, binding mode analysis, molecular dynamics, and prediction of ADMET/toxicity properties of selective potential antiviral agents against SARS-CoV-2 main protease: an effort toward drug repurposing to combat COVID-19. Mol Divers. 2021;25(3):1905-1927. https://doi.org/10.1007/s11030-021-10188-5 [ Links ]
70. Ishola AA, Joshi T, Abdulai SI, Tijjani H, Pundir H, Chandra S. Molecular basis for the repurposing of histamine H2-receptor antagonist to treat COVID-19. J Biomol Struct Dyn. 2022;40(13):5785-5802. https://doi.org/110.1080/07391102.2021.1873191 [ Links ]
71. Ahamad S, Gupta D, Kumar V. Targeting SARS-CoV-2 nucleocapsid oligomerization: insights from molecular docking and molecular dynamics simulations. J Biomol Struct Dyn. 2022;40(6):2430-2443. https://doi.org/10.1080/07391102.2020.1839563 [ Links ]
72. Milenkovic DA, Dimic DS, Avdovic EH, Markovic ZS. Several coumarin derivatives and their Pd (II) complexes as potential inhibitors of the main protease of SARS-CoV-2, an in silico approach. RSC Advances. 2020;10(58):35099-35108. https://doi.org/10.1039/D0RA07062A [ Links ]
73. Liu H, Hou T. CaFE: a tool for binding affinity prediction using end-point free energy methods. Bioinformatics. 2016;32(14):2216-2218. https://doi.org/10.1093/bioinformatics/btw215 [ Links ]
74. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33(12):889-897. https://doi.org/10.1021/ar000033j [ Links ]
75. Ibrahim MT, Uzairu A, Uba S, Shallangwa GA. Computational virtual screening and structure-based design of some epidermal growth factor receptor inhibitors. Future J Pharm Sci. 2020;6:1-16. [ Links ]
76. Lynch T, Price AL. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76:391-396. [ Links ]
77. Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000;38(1):41-57. https://doi.org/10.2165/00003088-200038010-00003 [ Links ]
Received 09 December 2021
Revised 14 June 2022
Accepted 19 July 2022
* To whom correspondence should be addressed: Email: mshabbir@kku.edu.sa; avedkhattak79@gmail.com

