










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Chemistry
versão On-line ISSN 1996-840X
versão impressa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.74 Durban 2021
http://dx.doi.org/10.17159/0379-4350/2021/v74a5
RESEARCH ARTICLE
Approaching Bulk from the Nanoscale: Extrapolation of Binding Energy from Rock-Salt Cuts of Alkaline Earth Metal Oxides
Susanne G.E.T. Escher; Helen D. Duncan*; Alexey A. Sokol; Scott M. Woodley
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
ABSTRACT
A systematic DFT study is performed on (MgO)B, (CaO)n, (SrO)n, and (BaO)n clusters with 6 < n < 50, and which display a cuboid 2X2X2 atomic motif seen in the bulk, rock-salt, configuration. The stability and energy progression of these clusters are used to predict the energies of infinitely long nanorods, or nanowires, slabs, and the bulk global minimum energy.
Keywords: Alkaline earth metal oxides, nanoclusters, nanorods, DFT.
1. Introduction
Chemistry typically takes place at the surface of a material and employing a nanoparticle form of a material beneficially increases the surface area to volume ratio. For even smaller particles, so-called nanoclusters, the atomic structure and therefore their properties would change dramatically with size. Given that an experimental technique is yet to be developed that can directly determine the atomic structure of nanoclusters, in the same way Bragg scattering, or X-ray diffraction experiments can be employed for sufficiently sized crystals. Research on nano-clusters is reliant upon computational predictive studies.1
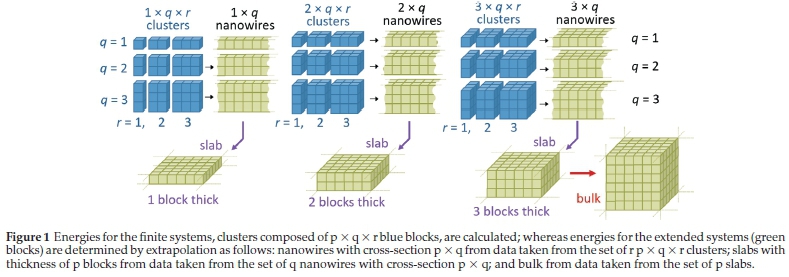
Experimentally observed crystal growth from nanoclusters to nanoparticles and beyond to bulk may proceed along different routes with nanostructures of diverse architecture reported in the literature that range from small bulklike crystallites to platelets, rods, tripods and flowers, wires and tubes, ultrathin films and even one-atom-thick monolayers typically grown on supports.2 In practice, realization of specific morphologies is usually kinetically controlled. The thermodynamic stability of the nanostructured objects with respect to bulk and, more generally, the way bulk properties develop upon growth in finite systems are still not fully understood within microscopic theories (although some insights are obtained from semi-qualitative models, including, e.g., quantum confinement). In this work, we apply a systematic approach3 to the investigation of how the bulk energy of a compound of interest can be estimated from the energies of finite nanoscale systems, starting with nanoclusters forming nanorods, which are extended to nanowires; then assembling the nanowires in nanosheets that form ultrathin films of increasing thickness; and, finally, the films evolving into the bulk as illustrated in Fig. 1. Constructing nanowires from clusters is not a new idea3-4, and has been employed in the prediction of a new structure for zinc sulphide.5
Beyond the fundamental curiosity, the structure and property evolution in the nanostructures with size has been the focus of many studies aimed at determining the relative stability of different bulklike phases, for example, considering competition between hexagonal (wurtzite) and cubic (zinc blende) structures in nanoparticles of II-VI semiconductor compounds.2,4,6 On the other hand, within the modelling community there is a high interest in finding series of representative nanostructures of computationally manageable size that can be exploited to predict physical properties and reactivity of bulk, either directly or by extrapolation.7
Our first application of the proposed method is to alkaline earth metal oxides, the cuboid [1 0 0] cuts of which as shown by experiment resemble the shape of observed nanowires.8 We predict properties of nanowires and slabs that can be constructed from the smallest cuboid metal oxide (MO)4 nanocluster, which is in fact only metastable in the gas phase. Note, that properties of nanowires, or nanorods, and slabs have attracted interest9 due to applications in field emission devices10 and sensing.11
Early studies of nanoclusters typically reported the tentative global energy minimum atomic configuration of a compound as a function of the number of formula units, n, it is composed of. The structural motif of these global minima for a compound will also typically change with size n; the smallest global minima may not resemble cuts that can be taken from the crystal structure of the bulk phase. Nanoclusters of zinc oxide is a good example of this change in non-bulklike structural motifs: the smallest global minima are rings, bubbles, double bubbles.1,12 In contrast, many of the relatively more stable global minima for the smaller nanoclusters of alkaline earth oxides have cuboid atomic configurations and therefore resemble cuts from their bulk phase (the NaCl rock-salt structure).
Even if the stoichiometry is maintained, nanoclusters display a wide structural diversity. Take, for example, the relatively small (MgO)5 cluster, which is predicted to have five unique metastable structures.13 Given that the properties of materials are dependent on their atomic and electronic structure, nano-clusters can potentially display a diverse range of properties compared to their corresponding bulk.
Of the alkaline earth metal oxides (MO)n clusters, magnesium oxide clusters, which have applications in gas and photoactive14 sensing devices, have been extensively investigated.6a,1s Structural and electronic properties have been reported for (MgO)n clusters up to n = 40,6a,15c,15d,15f,:16 and for nanotubes up to n = 33.15b,15d,16For the smallest (n < 30) (MgO)n clusters, another structural motif was predicted to be that of the tentative global minima for atomic structures resembling hexagonal-barrels.13a
Conversely the global minima clusters of the Ca, Sr, and Ba analogues show a preference for rock-salt cuts at sizes as small as n = 4 and specifically a preference for cuboid rock-salt cuts persists at arbitrarily small sizes, even where barrel shapes, bubbles or non-cuboid rock-salt cuts are also local minima.17
An extrapolation approach has been previously employed for magnesium and calcium oxides using the normalized clustering energy (NCE) of the global minima in the cluster size range of n = 2-1016,18 to predict the energy at the bulk limit. In our early work on the stability of (MO)n clusters with n = 2-24, we found that while most of these clusters have rock-salt-like global minima, not all do,13a,17a and, therefore, the stability trends might have an undue noise that should hinder accurate prediction. But this problem can be solved by a judicious construction of cluster series using one building unit, i.e. following the same structural motif will be demonstrated below.
In this paper, we will investigate ever-larger atomic structures that can be constructed from the n = 4,2x2x2 cuboid building unit; first expanding in one, then in two, and finally in all three dimensions. These series of calculations will allow us to extrapolate the energies of a one-dimensional nanowire and a two-dimensional slab. This method will be applied to the whole series of stable alkaline earth metal oxides: MgO, CaO, SrO, and BaO. Our hypothesis is that extrapolation using data from clusters that do not include the non-cuboid global minima should yield a more accurate value of the bulk energy. We also extrapolate directly from the energy of cubic cuts to obtain an estimate of the bulk energy. In the following section we outline the methods used. After that we present our results along with a discussion and conclude with a summary of our findings.
2. Methods
2.1. Cluster Geometry Optimization
The initial unrelaxed structures for most cuboid structures for (MO)nn < 25 were taken from our previous data,13a,17a while all other clusters were generated for this study with the metal-oxygen bond length set to that found in the bulk.19 The geometry of all clusters was optimized in FHI-aims20 with the PBEsol21 GGA functional and tight settings for the density grid, Hartree potential, basis functions cut-offs and tolerances imposed on convergence. The geometry was considered converged when the maximum residual force component per atom was below 1 x 10-3 eV A-1. FHI-aims tier 1 basis functions was used for all cations, which is analogous to DNVP(1d1f), i.e. double numerical valence with polarization, whereas FHI-aims tier 2 basis functions for anions, which is analogous to TNVP(2d1f1g), i.e. triple numerical valence with enhanced polarization.
2.2. Cluster Band Gap Calculations
Single point calculations were performed on the optimized geometries using the PBEsol0 functional and a tight basis set. Tier 1 basis functions were used for all cations, and tier 2 for oxygen anions.
2.3. Calculation of the Normalized Clustering Energy
For all the cuboid clusters the Normalized Clustering Energy (NCE) was calculated. The NCE22 is a measure of how much more stable a cluster is compared to the n = 1 smallest stoichiometric global minimum configuration, i.e. the fully relaxed dimer molecule. For a total energy £(n) of a (MO)n cluster, and with the corresponding energy of the M-O dimer, E(1), the normalized clustering energy is
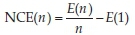
The NCE can be extended to one- two- and three-dimensional structures by taking n to be the number of formula units in the unit cell for which the energy is given; these energies will be referred to as generalized NCEs (gNCE).
2.4. Direct Calculation of the NCE for Periodic Structures
The energies of the 1D, 2D and 3D oxides were directly calculated using FHI-aims and the same functionals as for the geometry relaxation and employed periodic boundary conditions. For the one- and two-dimensional materials vacuum gaps were utilized to ensure that the separation between the materials and their periodic images was large enough that they did not interact with each other.
3. Results and Discussion
3.1. Building Blocks
This study focuses on clusters of the group 2 oxides in sizes ranging from2x2x3atoms, n =6,upton = 50 (all possible cuboids except the4x4x5 cuboid). Later a direct extrapolation of the bulk energy will be obtained from the energies of the series of perfect cube cuts where we will notice that a reasonable linear fit is hampered by the inclusion of the smallest cluster. Moreover, given that the composition of the2x2x2 clusters contains only three-coordinated corners atoms (see Fig. 2a), i.e. all atoms severely undercoordinated, the energy of this smallest cluster is not used in any of the three-step extrapolation. Figure 2 illustrates the nomenclature used within this paper; all cuboid clusters are defined in terms of Nxx Ny x Nz, where each number, N, is the number of atoms along the axis.
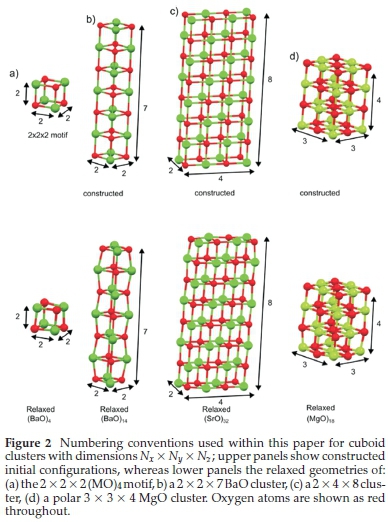
3.2. Clusters to Nanowires
Initially, the2x2x Nzclusters were considered, with the geometry optimized in FHI-aims. The NCE was calculated for Nz ranging from 3 to 25 (n = 6 to 50) and plotted as a function of the number of formula units, n, shown in Fig. 3.
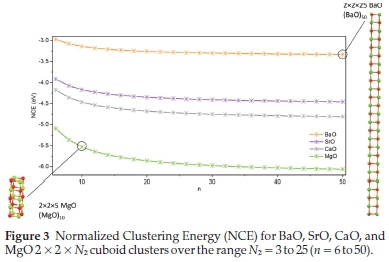
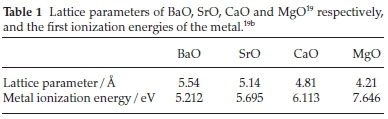
From Fig. 3 we see the NCE is lowest for MgO clusters and highest for BaO clusters, with the NCE for CaO, and SrO clusters approximately midway between. The similarity of NCE energy for Ca, and Sr clusters is expected due to their more similar sizes and ionization energies - shown in Table 1.
As expected, stabilizing effects increase with cluster size as well as decreasing cation size. In Fig. 3 we also observe an inverse relationship between cluster size and NCE. When the NCE is plotted as a function of 1/n, shown in Fig. 4 a linear fit to the data can be applied, which can be extrapolated to infinitely long nanowires, as 1/n tends to 0.
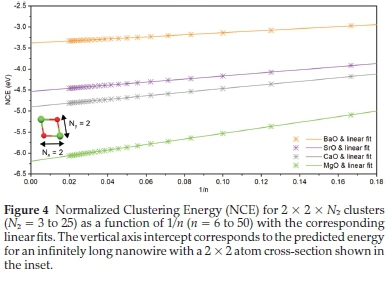
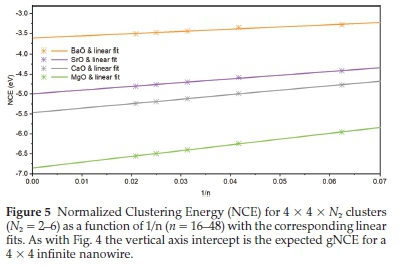
The vertical-axis intercepts of the linear fits from Fig. 4 are the predicted generalized NCE (gNCE) for infinitely long nano-wires with a 2 x 2 atom cross-section. Next we systematically increased the nanarods in transverse directions from Nx= 2 and Ny= 2, to Nx= 4 and Ny= 4, over a range of Nzvalues and the NCE calculated. Linear fits of the data made to predict the energy of nanowires with various cross-sections. For example, in Fig. 5, the NCE is plotted as a function of 1/n for clusters with a 4 x 4 atom cross-section, predicts nanowire energies that are lower than the corresponding 2 x 2 atom cross-section analogues.
The3x3x Nzdata set are limited as we have decided to only investigate charge-neutral clusters, so for this set only even values of Nz were considered. The3x3x Nz data are also expected - and found - to predict higher gNCEs than even-atom diameter nanowires. This is because all the clusters used to predict the gNCE are polar - see Fig. 2d - and are less stable (higher in energy) than their non-polar counterparts.17a Although barium is more polarizable than the smaller cations, this effect is most prominent in (BaO)n clusters and diminishes with a decrease in cation size. Table 2 contains the intercepts for the full range of clusters investigated, and their associated uncertainty.
From Table 2 we can see the predicted gNCE for an infinitely long SrO nanowire with a2x3 atom cross-section is -4.679(7) eV and that the uncertainty on the2x2 nanowires, for which the greatest number of unique clusters were analyzed, is smaller than all others.
3.3. Nanowires to Slabs
The extrapolated nanowire data from Table 2 can then be used to extrapolate data further to 2D structures namely the two-, three- and four-atom thick slabs, from two-, three- and four-atom width nanowires, and is shown in Fig. 6.
As with the cluster-to-wire calculation, extrapolation of a linear fit to the data is used to predict the gNCE for two-, three-, and four-atom thick slabs, given in Table 3.
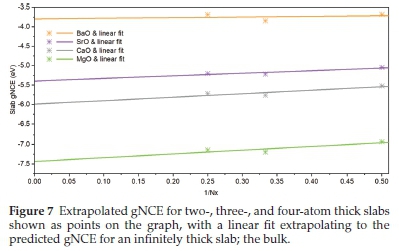
3.4. Extrapolation to the Bulk
The slab gNCEs can be used to predict the bulk energies; as shown in Fig. 7, the vertical axis intercepts now correspond to our predicted bulk gNCE for the group 2 oxides, which has been tabulated in Table 4 and compared to the energies derived using the same exchange and correlation density functionals for the 3D periodic structures.
We compared the extrapolated bulk gNCE using the wire to slab to bulk three-step method, to the predicted gNCE when all clusters are considered in one-step. The NCE for all cubic clusters produced in this work were plotted as a function of 1/n and fit in a one-step process, as shown in Fig. 8 with the extrapolated bulk gNCE also given in Table 4.
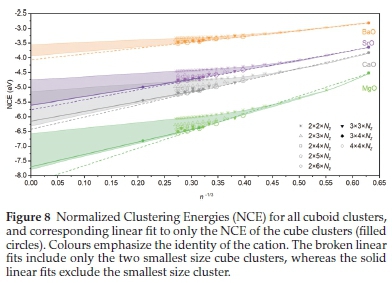
The three- and one-step methods predict energies which bracket the values resulting from DFT using periodic boundary conditions (PBC). Energies derived using the one-step approach that includes/excludes the smallest cube clusters are significantly further/nearer to the target PBC values. The three- and one-step methods are consistently higher and lower than the target energy. Moreover, the three- and one-step methods provide energies that are closer to the target values for smaller and larger cations, respectively.
3.5 HOMO-LUMO Gaps for 2 X 2 X NzClusters
Additional single-point calculations were performed on the optimized 2 x 2 x Nzset of clusters using FHI-aims with the PBEsolO functional. The overall HOMO-LUMO gap was calculated and is shown in Fig. 9b. In contrast to the bulk, the size of the HOMO-LUMO gap is not clearly correlated with cation size. To explain the observed trends, we recall that the magnesium ion has the largest difference in ionic radius as compared to the oxygen anion, which results in strong ionic relaxations away from ideal cubic lattice positions. Thus, a significant stabilization of the electronic levels for states localized on magnesium should be expected (polarization). This explains the significant drop in the position of the LUMO energies for MgO compared to the other three compounds - see Fig. 9a. The trends we observe in the HOMO energies, shown in Fig. 9a, are in qualitative agreement with experimentally derived and computational values of the ionization potentials for the respective bulk compounds.23
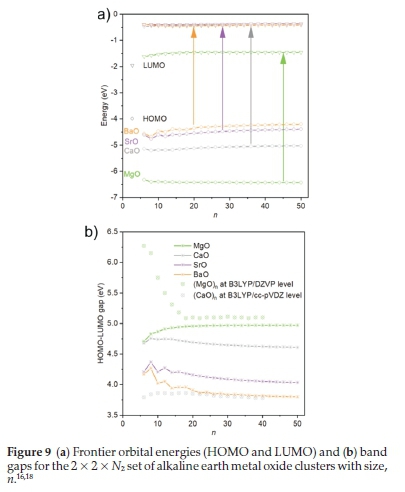
The LUMO energies are still below the vacuum level, as opposed to above for the bulk, which is clearly a finite size effect that again can be traced back to the polarization. Figure 9b shows our calculated values of the HOMO-LUMO gap compared to that of MgO 2x2x Nzclusters reported in I16! and CaO 2x2x Nzclusters reported in the t18!. While it can be noted that the trends in the HOMO-LUMO gaps for the smaller clusters are different in this work from published in I16,18l. The differing choice of DFT flavours, i.e. exchange and correlation functional (by necessity not exact) and choice of basis set (by necessity not complete), introduces errors, and highlights that any analysis here can only really touch on internal consistency.
The calculated HOMO-LUMO gaps can be related to the chemical hardness, calculated as Ionization potential - Electron Affinity)/2. From Fig. 9b, it appears that the chemical hardness is dependent upon the size of the cation, i.e. the larger the cation the lower the hardness. For the chemically softer structures, the chemical hardness reduces with the length of the rods. In fact, strontium oxide is more similar to barium oxide than calcium oxide, which perhaps explains why the interatomic distances and lattice parameters of strontium oxide structures are closer to those of calcium oxide.
4. Conclusion
In this paper we present a systematic study of group two oxide clusters with cuboid atomic motifs. By calculation of the Normalized Clustering Energy (NCE), the generalized NCE (gNCE) for infinitely long nanowires with cross-sections varying from 2 x 2 to4x4 atoms were predicted. Similarities in the NCE, and hence the gNCE for Ca and Sr clusters arise as the dominant contribution to the energy comes from the interatomic Coulomb term, which is dependent upon interatomic distance (cation size) and ionization energies. The nanowire-predicted gNCEs were used to predict the gNCE for slabs of two-, three-, and four-atom thickness. The NCE for all clusters were used to predict the bulk (periodic) gNCE via a one-step and three-step method. The predicted gNCE bracket the observed periodic calculated values. gNCE predicted using the one-step method where the smallest clusters were excluded (method B) were closer to target energy than predictions from the one-step method that included smaller cations. The three-step method (nanowire-slab-bulk) predicts gNCEs closer to the target energies for smaller cations, whereas the one-step method shows a closer agreement to target energies for larger cations. Analysis of the HOMO-LUMO gaps for the2x2x Nzclusters shows the chemical hardness is dependent on cation size, with SrO and BaO clusters displaying greatest similarity, and smaller cations displaying greater hardness.
Having shown that three- and one-step procedures can be employed to obtain an upper and lower bound on the bulk energy typically calculated using periodic boundary conditions, it may prove to be a useful procedure for other classes of chemical substances and topologies.
Acknowledgements
We are grateful to UCL for providing a DTA studentship and EPSRC for the WASP@N and SAINT grants (EP/I03014X and EP/P022235). Through our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/R029431), this work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk) and the UK Materials and Molecular Modelling Hub for computational resources, MMM Hub, which is partially funded by EPSRC (EP/P020194). Additionally, we acknowledge the use of the WASP@N, or Hive, database13b where we will also upload atomic structures of clusters from this report. We thank Volker Blum who guided us with our FHI-aims calculations and Martijn Zwijnenburg for useful discussions.
Author Contributions
Susanne G.E.T. Escher and Helen D. Duncan carried out the calculations and analysis; Scott M. Woodley created Fig. 1 and with Alexey A. Sokol provided leadership and steered the project.
ORCID iDs
S.G.E.T. Escher: orcid.org/0000-0002-4825-729X
H.D. Duncan: orcid.org/0000-0002-0897-7188
A.A. Sokol: orcid.org/0000-0003-0178-1147
S.M. Woodley: orcid.org/0000-0003-3418-9043
References
1 S.M. Woodley and S.T. Bromley, in Frontiers of Nanoscience, (S.T. Bromley and S.M. Woodley, eds.), vol. 12, Elsevier, 2018, pp. 1-54. [ Links ]
2 S. Hamad, S.M. Woodley and C.R.A. Catlow, Experimental and computational studies of ZnS nanostructures, Mol. Simulat., 2009, 35, 1015-1032. [ Links ]
3 M.Y. Chen and D.A. Dixon, Modeling the formation of TiO2 ultra-small nanoparticles, Nanoscale, 2017, 9, 7143-7162. [ Links ]
4 C.R.A. Catlow, S.A. French, A.A. Sokol, A.A. Al-Sunaidi and S.M. Woodley, Zinc oxide: a case study in contemporary computational solid state chemistry, J. Comput. Chem., 2008, 29, 2234-2249. [ Links ]
5 N. Krainara, J. Limtrakul, F. Illas and S.T. Bromley, Magic numbers in a one-dimensional nanosystem: ZnS single-wallednanotubes, J. Phys. Chem. C, 2013,117, 22908-22914. [ Links ]
6 a) C. Roberts and R.L. Johnston, Investigation of the structures of MgO clusters using a genetic algorithm, Phys. Chem. Chem. Phys., 2001, 3, 5024-5034; b) C.R.A. Catlow, S.T. Bromley, S. Hamad, M. Mora-Fonz, A.A. Sokol and S.M. Woodley, Modelling nano-clusters and nucleation, Phys. Chem. Chem. Phys., 2010,12, 786-811. [ Links ]
7 a) O. Lamiel-Garcia, A. Cuko, M. Calatayud, F. Illas and S.T. Bromley, Predicting size-dependent emergence of crystallinity in nano-materials: titania nanoclusters versus nanocrystals, Nanoscale, 2017,9, 1049-1058; b) S.T. Bromley, I.D.R. Moreira, K.M. Neyman and F. Illas, Approaching nanoscale oxides: models and theoretical methods, Chem. Soc. Rev., 2009, 38, 2657-2670. [ Links ]
8 C.C. Tang, Y. Bando and T. Sato, Oxide-assisted catalytic growth of MgO nanowires with uniform diameter distribution, J. Phys. Chem. B, 2002,106, 7449-7452. [ Links ]
9 A.A. Al-Ghamdi, F. Al-Hazmi, F. Alnowaiser, R.M. Al-Tuwirqi, A.A. Al-Ghamdi, O.A. Alhartomy, F. El-Tantawy and F. Yakuphanoglu, A new facile synthesis of ultra fine magnesium oxide nanowires and optical properties, J. Electroceram., 2012, 29, 198-203. [ Links ]
10 Y.K. Cui, J. Chen, X.B. Zhang, W. Lei, Y.S. Di and Q.L. Wang, Flexible field emission devices based on barium oxide nanowires, J. Disp. Technol, 2016,12, 466-471. [ Links ]
11 C.H. Kao, C.L. Chang, W.M. Su, Y.T. Chen, C.C. Lu, Y.S. Lee, C.H. Hong, C.Y. Lin and H. Chen, Magnesium oxide (MgO) pH-sensitive sensing membrane in electrolyte-insulator-semiconductor structures with CF4 plasma treatment, Sci. Rep-UK, 2017, 7. [ Links ]
12 A.A. Al-Sunaidi, A.A. Sokol, C.R.A. Catlow and S.M. Woodley, Structures of zinc oxide nanoclusters: as found by revolutionary algorithm techniques, J. Phys. Chem. C, 2008, 112, 18860-18875. [ Links ]
13 a) S.G.E.T. Escher, T. Lazauskas, M.A. Zwijnenburg and S.M. Woodley, Synthesis target structures for alkaline earth oxide clusters, Inorganics, 2018, 6;b) University College London, WASP@N - Web Assisted Structure Prediction at the Nanoscale. https://hive.chem.ucl.ac.uW [ Links ]
14 A. Sternig, S. Stankic, M. Muller, J. Bernardi, E. Knozinger and O. Diwald, Photoluminescent nanoparticle surfaces: the potential of alkaline earth oxides for optical applications, Adv. Mater., 2008, 20, 4840-4844. [ Links ]
15 a) J.V Stark, D. G. Park, I. Lagadic and K.J. Klabunde, Nanoscale metal oxide particles/clusters as chemical reagents. Unique surface chemistry on magnesium oxide as shown by enhanced adsorption of acid gases (sulfur dioxide and carbon dioxide) and pressure dependence, Chem. Mater., 1996, 8, 1904-1912; b) G. Bilalbegovic, Structural and electronic properties of MgO nanotube clusters, Phys. Rev. B, 2004,70; c) K. Kwapien, M. Sierka, J. Dobler, J. Sauer, M. Haertelt, A. Fielicke and G. Meijer, Structural diversity and flexibility of MgO gas-phase clusters, Angew Chem. Int. Ed., 2011, 50,1716-1719; d) M. Haertelt, A. Fielicke, G. Meijer, K. Kwapien, M. Sierka and J. Sauer, Structure determination of neutral MgO clusters-hexagonal nanotubes and cages, Phys. Chem. Chem. Phys.,2012,14,2849-2856; e) M.C.C. Wobbe, A. Kerridge and M.A. Zwijnenburg Optical excitation of MgO nanoparticles; a computational perspective, Phys. Chem. Chem. Phys., 2014,16, 22052-22061; f) M.R. Farrow, Y. Chow and S.M. Woodley, Structure prediction of nanoclusters; a direct or a pre-screened search on the DFT energy landscape?, Phys. Chem. Chem. Phys., 2014, 16, 21119-21134. [ Links ]
16 M.Y. Chen, A.R. Felmy and D.A. Dixon, Structures and stabilities of (MgO)(n) nanoclusters, J. Phys. Chem. A, 2014, 118, 3136-3146. [ Links ]
17 a) S.G.E.T. Escher, T. Lazauskas, M.A. Zwijnenburg and S.M. Woodley, Structure prediction of (BaO)(n) nanoclusters for n < = 24 evolutionary algorithm, Comput. Theor. Chem., 2017,1107,74-81; b) F. Bawa and I. Panas, Competing pathways for MgO, CaO, SrO, and BaO nanocluster growth, Phys. Chem. Chem. Phys., 2002, 4, 103-108; c) P. Batra, R. Gaba, U. Issar and R. Kakkar, Structures and stabilities of alkaline earth metal oxide nanoclusters: a DFT study, 2013. [ Links ]
18 M.Y. Chen, K.S. Thanthiriwatte and D.A. Dixon, Structures and stabilities of (CaO)(n) nanoclusters, J. Phys. Chem. C, 2017, 121, 23025-23038. [ Links ]
19 a) W. Primak, H. Kaufman and R. Ward, X-Ray Diffraction studies of systems involved in the preparation of alkaline earth sulfide and selenide phosphors, J. Am. Chem. Soc., 1948, 70, 2043-2046; b) D.R. Lide, CRC Handbook of Chemistry and Physics, 89th edn., CRC Press, Boca Raton, FL, USA, 2009; c) R.J. Zollweg, X-Ray lattice constant of barium oxide, Phys. Rev., 1955, 100, 671-673. [ Links ]
20 V. Blum, R. Gehrke, F. Hanke, P. Havu, V Havu, X.G. Ren, K. Reuter and M. Scheffler, Ab initio molecular simulations with numeric atom-centered orbitals, Comput. Phys. Commun., 2009,180,2175-2196. [ Links ]
21 a) J.P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865-3868; b) J.P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1997, 78, 1396-1396; c) J.P. Perdew, A. Ruzsinszky, G.I. Csonka, O.A. Vydrov, G.E. Scuseria, L.A. Constantin, X.L. Zhou and K. Burke, Restoring the density-gradient expansion for exchange in solids and surfaces, Phys. Rev. Lett., 2008, 100. [ Links ]
22 D. Labanc, M. Sulka, M. Pitoñák, I. éernusák, M. Urban and P Neogrády, Benchmark CCSD(T) and DFT study of binding energies in Be7 -12: in search of reliable DFT functional for beryllium clusters, Mol. Phys., 2018, 116, 1259-1274. [ Links ]
23 A.J. Logsdail, D.O. Scanlon, C.R.A. Catlow and A.A. Sokol, Bulk ionization potentials and band alignments from three-dimensional periodic calculations as demonstrated on rocksalt oxides, Phys. Rev. B, 2014, 90. [ Links ]
Received 17 April 2020
Revised 13 May 2021
Accepted 10 June 2021
* To whom correspondence should be addressed. E-mail: h.duncan@ucl.ac.uk


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}