










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.74 Durban 2021
http://dx.doi.org/10.17159/0379-4350/2021/v74a10
RESEARCH ARTICLE
Competitive Adsorption of H2O and SO2 on Catalytic Platinum Surfaces: a Density Functional Theory Study
Marietjie J. UngererI, II; David Santos-CarballalII, III; Cornelia G.C.E. van SittertI,*; Nora H. de LeeuwII, III, IV,*
ILaboratory for Applied Molecular Modelling, Research Focus Area: Chemical Resource Beneficiation, North-West University, Private Bag X6001, Potchefstroom, 2520, South Africa
IISchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, United Kingdom
IIISchool of Chemistry, University of Leeds, Leeds LS2 9JT, United Kingdom
IVDepartment of Earth Sciences, Utrecht University, Princetonplein 8A, 3584 CD Utrecht, Netherlands
ABSTRACT
Platinum has been widely used as the catalyst of choice for the production of hydrogen in the hybrid sulphur (HyS) cycle. In this cycle, water (H2O) and sulphur dioxide (SO2) react to form sulphuric acid and hydrogen. However, the surface reactivity of platinum towards H2O and SO2 is not yet fully understood, especially considering the competitive adsorption that may occur on the surface. In this study, we have carried out density functional theory calculations with long-range dispersion corrections [DFT-D3-(BJ)] to investigate the competitive effect of both H2O and SO2 on the Pt (001), (011) and (111) surfaces. Comparing the adsorption of a single H2O molecule on the various Pt surfaces, it was found that the lowest adsorption energy (Eads = -1.758 eV) was obtained for the dissociative adsorption of H2O on the (001) surface, followed by the molecular adsorption on the (011) surface (Eads = -0.699 eV) and (111) surface (Eads = -0.464 eV). For the molecular SO2 adsorption, the trend was similar, with the lowest adsorption energy (Eads = -2.471 eV) obtained on the (001) surface, followed by the (011) surface (Eads = -2.390 eV) and (111) surface (Eads = -1.852 eV). During competitive adsorption by H2O and SO2, the SO2 molecule will therefore preferentially adsorb onto the Pt surface. If the concentration of SO2 increases, self-reaction between two neighbouring SO2 molecules may occur, leading to the formation of sulphur monoxide (SO) and -trioxide (SO3) on the surface, which could lead to sulphur poisoning of the Pt catalytic surface.
Keywords: Platinum, water, sulphur dioxide, hydrogen, adsorption, density functional theory.
1. Introduction
The increasing demand to reduce toxic emissions, not only from automotive engines,1 but also for example in the aviation sector,2 steel manufacturing and electricity generation,3 has driven extensive research toward the production of clean, renewable and sustainable energy, e.g. from wind,4,5 solar,6-8hydroelectricity9,10 or combinations thereof,11-13 or alternative energy sources such as hydrogen (H2).14 Currently, various different feedstocks are used for the production of H2,15 including from biomass,16 nuclear17 or waste water,18 the non-carbon-based hybrid sulphur (HyS) cycle which has shown itself as a promising, potentially large-scale process.19,20 During the HyS cycle, sulphur dioxide (SO2)/sulphuric acid (H2SO4) is used in an electro-oxidation reaction, leading to the net reaction of splitting the water (H2O) into H2 and oxygen (O2). Various anode catalysts20 have been tested for this reaction and the carbon supported metallic platinum (Pt) catalyst has consistently shown both high activity and stability21-23 towards this reaction.
Sulphur-containing molecules, including SO2, SO and S, have long been known to be among the key poisoning compounds in heterogeneous catalysis.24 As such, one of the major problems with Pt anode catalysts is the formation of a sulphur (S) layer on the surface, leading to catalyst poisoning, i.e. deactivation and in severe cases surface delamination.25,26 Despite their acknowledged role in the poisoning of these supported Pt metal catalysts,26 the fundamental chemistry and mechanistic behaviour of the sulphur-metal interaction remains poorly defined. To understand the deactivation mechanism on a fundamental level, some experimental and theoretical studies focussed on SO2 adsorption on various Miller indexes of pure metal surfaces, including Cu,27-30 Ni31-33 Ag,34,35 Rh,36,37 Pd29,37-41 and Pt29,36,42-45.
Although Pt is the most studied system, opposing results have been obtained for SO2 adsorbed onto the Pt (111) surface.24 This is due to the operational conditions, e.g. surface coverage and surface morphology, during the SO2 oxidation/reduction reactions, influencing the thermodynamics leading to different final products.46 However, major difficulties have been experienced in experiments, because even when pure sulphur oxides, such as SO2, were adsorbed from the gas phase onto single metallic catalysts, a number of co-adsorbed sulphur species were detected on the surface.47-49 Moreover, very little theoretical work or modelling has been performed on evaluating the reaction energies and thermodynamics of these sulphur oxides with the various Pt surfaces.
Pt is widely used in various reactions in which water acts both as a participant or bystander,50 influencing the behaviour of the heterogeneous catalytic surface.51 The nature of the H2O-metal interaction is of obvious importance52,53 and considerable research effort has been devoted towards understanding these systems.54-56 A major challenge in modelling the adsorption of water on a catalytic surface are the multitude of atomic position variations in the simulated liquid, which necessitates the addition of several different configurations in the initial computational set up. Previous modelling studies showed58,59 that the most reliable results extensively looked at the way the water molecules interacts with each other and the surface and does not necessitate adsorption of additional water molecules onto the surface. Another factor to consider is the splitting of the water molecule into H+ + OH- and how these species affect not only the catalytic surface, but also the behaviour of co-adsorbed species. It is evident that the detailed description of the binding behaviour of water molecules onto Pt surfaces is still not complete. The starting point here will be an in-depth understanding of the behaviour between the H2O molecules and the metal surface atoms.
In this paper, we have used DFT calculations to predict the behaviour of H2O and SO2 on the electro-catalytically active surfaces of platinum metal, i.e. the Pt (001), (011) and (111) surfaces. We examine the adsorption energy of various geometries, any charge transfer between the Pt surface and the adsorbates as well as the work function. The overall aim of our study was the development of a comprehensive understanding of the H2O-surface and SO2-surface chemistry on the electro-catalytically active surfaces of Pt, and in particular the competition between these two reactants in the HyS cycle for specific surface adsorption sites, which will be the initial step in the overall HyS reaction process.
2. Computational Methods
2.1. Surface Calculation Details
The Vienna Ab Initio Simulation Package (VASP)60-63 has been shown to give accurate surface adsorption data64-67 and was therefore used to simulate the Pt surfaces and their interactions with H2O68 and SO2.69 The projector augmented wave (PAW)70,71 method was employed to describe the interaction between the valence and the core electrons. The core electrons of Pt were defined up to and including the 5p orbitals. The Perdew, Burke and Ernzerhof (PBE)72,73 functional within the generalized gradient approximation (GGA) was applied in all calculations. Plane-waves were included to a cut-off of 400 eV. The long-range dispersion interactions were considered with the DFT-D3 method with Becke-Johnson (BJ) damping.74 The Methfessel-Paxton scheme order 175 was used with a smearing of 0.05 eV to determine the partial occupancies during geometry optimization, ensuring an electronic entropy of less than 1 meV atom-1. However, the tetrahedron method with Blöchl corrections76 was used in the final static simulations to obtain accurate total energies and charges. The electronic and ionic optimization criteria were 10-5 eV and 10-2 eV A-1, respectively, and the conjugate gradient technique was adopted for the geometry optimizations.
Pt has a Fm3m crystal structure77 and the bulk Pt structure was calculated within a primitive face-centred cubic (fcc) cell using a r-centred 17 x 17 x 17 Monkhorst-Pack78 k-point mesh. Previous work has shown that long-range dispersion approximations influence not only the lattice parameters of a modelled surface, but also its surface energies.64,68 In this paper the geometry optimization of the Pt (001), (011) and (111) surfaces have therefore been carried out with the DFT-D3(BJ) method.74 Our calculated fcc Pt lattice constant was 3.926 A, which is in excellent agreement with the experimental value of 3.924 A.79,80
The Pt (001), (011) and (111) surfaces were investigated by simulating the periodic p(3 x 3), p(3 x 3) and p(4 x 4) supercells, respectively, which were generated from the bulk using the METADISE code.81 A vacuum of 15 A was added in the z-direc-tion perpendicular to the plane of the surface, to avoid interaction between the neighbouring cells. Each slab contained four atomic layers and the surface areas of the supercells were 138.17 A2,196.18 A2 and 106.79 A2 for the (001), (011) and (111) surfaces, respectively. The atoms in the two bottom layers of the slabs were fixed in the optimized bulk positions and the atoms in the remaining two layers were allowed to relax freely. A r-centred 7x7x1 Monkhorst-Pack fc-point grid was used for all the surface systems to sample the Brillouin zone.
The unrelaxed (γu) and relaxed (γr) surface energies were determined using Equations (1) and (2), respectively:
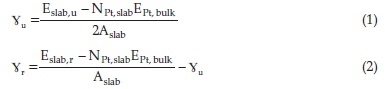
where Eslab,u,Eslab,r and EPtbulk,u are the energies of the unrelaxed slab, the half-relaxed slab and the bulk, respectively. NPt,slab and Aslab represent the number of Pt atoms in the slab and the surface area of the slab, respectively. The percentage relaxation (R) was calculated as the difference between the unrelaxed and relaxed surface energies, divided by the unrelaxed surface energy and multiplied by 100.
The work function (φ) is the minimum energy needed to remove an electron from the bulk of a material through a surface to a point outside the material. Here, we have calculated the energy needed to remove an electron from the Fermi level (Ef)of the metal surface to the vacuum potential (Evac) at 0 K.82
Atomic charges for the pristine surfaces were obtained using Bader analysis,83-86 which partitions space into non-spherical atomic regions enclosed by local minima in the charge density.
2.2. Adsorption Calculation Details
The isolated H2O and SO2 molecules for reference were optimized in periodic boxes of 12 x 13 x 14 A3 to ensure negligible interaction with their images in periodically repeated neighbouring cells. The Gaussian smearing scheme75 was used during geometry optimization and energy calculations were carried out with a smearing of 0.05 eV. A r-centred1x1x1 Monkhorst-Pack78fc-point mesh was used. Dipole corrections were added in all directions and the H2O and SO2 molecules were computed without symmetry. The core electrons of O and S were defined up to and including the 1s and 2p orbitals, respectively. For the H atoms, all the electrons were treated as valence electrons. Again, the atomic charges for the single molecules and adsorbed systems were obtained using Bader analysis.83-86
3. Results and Discussion
In this section we briefly describe the Pt surface slabs with the low Miller indices (001), (011) and (111) (Section 3.1), followed by an overview of the adsorption of both H2O and SO2 (Section 3.2), with detailed descriptions of their behaviour on the Pt (001), (011) and (111) surfaces discussed in Sections 3.2.1, 3.2.2 and 3.2.3, respectively.
3.1. Surface Structures
The top and side views of the Pt (001), (011) and (111) surfaces used in our simulations are shown in Fig. 1. To distinguish between top layer and subsequent layer atoms, the colour of the atoms in the top layer of each of the surfaces were changed to lighter silver. All three surfaces are planar, bulk-terminated structures, with each slab containing four atomic layers plus a 15 A vacuum space in the z-direction. The Pt (001) and Pt (111) surfaces are smooth with a face-centred cubic arrangement, while the Pt (011) is open-facetted, forming grooves on the surface. The adsorption sites indicated in Fig. 1 for the Pt (001) and (011) surfaces are atop (A), bridge (B) and four-fold hollow (4F), while the Pt (111) surface has atop (A), bridge (B), hexagonal close packed (hcp) and face-centred cubic (fcc) sites.
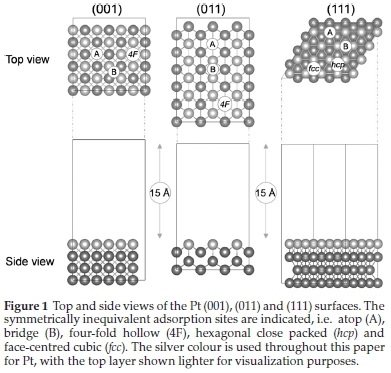
Table 1 lists the relaxed and unrelaxed surface energies and the surface areas for the Pt (001), (011) and (111) surfaces. In terms of surface energy, our calculations correlated with previously identified trends, where Pt (111) has the lowest surface energy and is hence the most stable plane, followed by the (001) and (011) surfaces. Literature reported an experimental surface energy of 2.48 J m-287 which is in good (quantitative) agreement with our calculated surface energies, particularly if we keep in mind that our perfect surfaces will lead to smaller surface energies than experimental surfaces, which are bound to contain defects that raise the surface energy.88
To understand the possible behaviour and chemical reactivity of the Pt (001), (011) and (111) surfaces, the work function (φ), was calculated for each pristine surface (Table 1). From our calculations, it can be seen that removing an electron would be easiest from the (001) surface, followed by the (111) and (011) surfaces. Literature showed a similar trend89 φ(011) < φ(001) < φ(111), with the lowest work function calculated for the (011) surface, followed by the (001) and (111) surfaces. However, the surface area and modelling approximation used have an effect on these values. Likewise will the surface properties and the temperature influence the work function data, which in its isolation cannot be used to predict reactivity.94
Previously, it has been shown that adsorption tendencies on transition metal surfaces correlate with the positions of the d-band centre.95 The overall tendency is that the higher in energy the occupied d-states, the stronger the bond with a molecule that accepts electrons from the metal. From our calculations it was seen that the Pt (111) surface had the highest d-band centre energy, followed by the (001) and (011) surfaces. Literature reported93 a d-band centre value of -2.45 eV for the Pt (111) surface, which is in excellent agreement with our calculations.
3.2. Adsorption of H2O and SO2
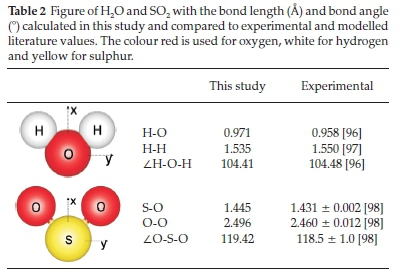
To calculate the adsorption behaviour of H2O on a Pt surface, a single H2O molecule in a box was modelled, shown in Table 2. The calculated H-O and H-H bond lengths deviated from the experimental gas phase values by less than 0.02 A and the H-O-H bond angle deviated by only ±0.07 °. Similarly, a single SO2 molecule in a box was modelled and shown in Table 2. The calculated S-O and O-O bond lengths compared to experimental gas phase values to within ±0.024 A and the O-S-O bond angle deviated by only ~1 °.
As shown in Fig. 1, various possible adsorption sites for both H2O and SO2 were considered on each surface. Adsorbed H2O molecules on metal surfaces is usually considered to be intact, except when co-adsorbed with other molecules or atoms.99,100 However, one study investigated a water bilayer on Ru (0001) and suggested that up to half the water molecules are dissociated, with one O-H bond broken in the dissociated water molecules.101 Similarly, up to9%ofthe H2O molecules dissociated in a study of water bilayers on Pt surfaces.58 Allowing that it would be less likely to have a single molecule of water dissociate on the surface, it was still decided to include these data on all three Pt surfaces for reasons of comparison.
Different H2O adsorption modes102 were considered on each site, including (i) where all three atoms of the H2O molecule is parallel to the Pt surface and could interact with the surface, (ii) where the oxygen was bound atop the Pt surface with both H atoms directed away from the surface, (iii) where OH was in the plane of the surface to interact and H was turned upward, and (iv) where one of the H atoms was turned downward to interact with the Pt surface. Five different SO2 adsorption modes were investigated on each Pt surface, i.e. (i) parallel, (ii) co-planar, (iii) bridging, (iv) O-bonded and (v) S,O-bonded.103 All five modes were investigated in the various adsorption sites shown in Fig. 1. The most favourable adsorption modes will be discussed for each of the Pt (001), (011) and (111) surfaces.
3.2.1. Pt (001)
The most stable and favourable adsorption modes of H2O and SO2 on the (001) surface found are shown in Fig. 2, with their calculated bond distances and angles of the adsorbed molecules listed in Table 3.
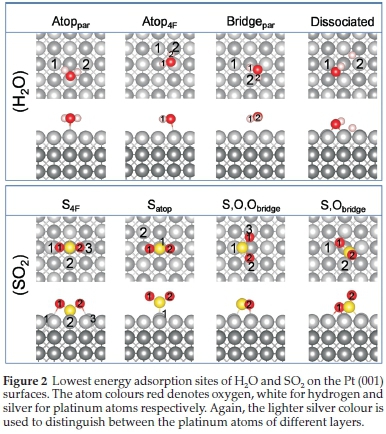
On the (001) surface, four very different H2O adsorption configurations were observed, i.e. three molecular adsorptions, Atoppar, Atop4F and Bridgepar, and one dissociated configuration, (001)diss. In the first mode of adsorption, Atoppar, the H2O molecule was parallel to the Pt surface with the H-atoms directed toward atop Pt atoms (Fig. 2). Here the H-O bond lengths and H-O-H bond angle were similar to the isolated H2O molecule (Table 2), indicating physisorption to the Pt surface. Similarly, in the second adsorption mode, Atop4F, the H2O molecule was also parallel to the Pt surface with the H-atoms directed toward the four-fold hollow position, where the O-Pt distance was 2.311 A and the H2-Pt distances were 2.831 and 2.786 AforPt1 and Pt2, respectively. The H-O-H angle correlated with experimental values at 104.48 °,104 again suggesting that the water was physisorbed. The third adsorption mode, Bridgepar, showed the O atom of H2O bound between two atop Pt atoms, with the H atoms directed toward a 4F hollow. However, the H2O molecule is not symmetrically parallel to the surface, with a Ptsurface-O-H1 bond angle of ~8 ° and a Ptsurface-O-H2 angle of ~3 °. Similar to the other two adsorption modes, we found that the O-H bond lengths and H-O-H bond angle correspond to the free molecule, indicating physisorption of the H2O molecule. In the case of (001)diss, both the OH and dissociated H atom were bound in the four-fold hollow site, with an O-Pt distance of 2.096 A and hydroxy H to Pt1 distance of 2.531 A and 2.956 A to Pt2. The H-Pt2 distance for the dissociated H was 1.754 A.
The calculated adsorption energies are tabulated in Table 3, which shows that the H2O molecule is much more strongly bound to surface when it is dissociated, whereas molecular adsorption follows the trend Atoppar > Bridgepar > Atop4F.Inthe matter of the dissociated H2O, we note that a charge of 0.39 e-was transferred from the Pt surface to the molecule, with the dissociated H atom becoming electron-depleted (Aq = 0.623 e-) relative to the surrounding Pt atoms, whereas the OH part gained electrons (Aq = -1.016 e-). However, in the molecular adsorption on the (001) surface, between 0.5 and 0.11 e- were donated from the molecule to the surface, and, as also suggested by the positive Aq values, the charge transfer values follow the same trend as the adsorption energies, except in the case of Atop4F. From our calculations, it appears that the H2O molecule would start in the Atoppar configuration (-1.675 eV, 0.109 e-), from where it has to move to the Atop4F configuration (-0.510 eV, 0.107 e-), with a lower adsorption energy but similar transferred charge, before it dissociates.
During the SO2 adsorption on the (001) surface, four possible adsorption modes were observed and named according to the adsorption site, i.e. S4F,Satop, S,O,Obridge and S,Obridge. In the first adsorption mode, S4F, the S atom is within a 4F hollow, bound to two surrounding Pt atoms and the two oxygen atoms are bound to the other two surrounding Pt atoms of the same 4F hollow. The S-O bond length is slightly elongated, while the O-S-O bond angle is smaller than for the free SO2 molecule, which indicates chemisorption on the (001) surface. The second adsorption mode is Satop, where the S atom is bound atop a Pt atom, with the O atoms directed away from the surface. In this case the S-O bond length and O-S-O bond angle correlate with the free SO2 molecule, because there is limited interaction between the surface and the adsorbed molecule. In the third adsorption mode, i.e. S,O,Obridge, the SO2 molecule is parallel to the Pt surface, with both O atoms bound to Pt surface atoms. Similar to the S4F configuration, the S-O bonds are elongated, while the O-S-O bond angle is smaller, again indicating chemisorption in this configuration. In the fourth adsorption mode, i.e. S,Obridge, one S-O bond is parallel to the surface, thereby binding to four Pt atoms in a 4F binding site, with the other O atom, O2, directed away from the surface. The S-O2 bond length correlates with the S-O bond length of the free SO2 molecule, while the S-O1 bond length is elongated due to the attraction to two Pt atoms in the 4F hollow site. In an experimental study of SO2 adsorption on a Pd (100) surface,38 SO2 had a S,Obridge geometry with a corresponding S-O and S-Pd bond length of 1.48 and 2.24 A, respectively. In an SO2 adsorption study on Ru (001),105 it was found that the molecular plane of SO2 was perpendicular to the Ru(001) surface, similar to the Satop and S4F adsorptions here, with a corresponding adsorption energy of 0.538 eV (12.4 kcal/mol). In another study on Cu (100),30 it was found that at low coverages SO2 should adsorb preferentially with its molecular plane parallel to the surface, similar to our S,O,Obridge adsorption. However, as the coverage of SO2 on Cu (100) becomes substantial, the molecule adopts the S,Obridge binding configurations to minimize adsorbate-adsorbate repulsions.
Comparing the adsorption energies of all four SO2 adsorption modes, we note that the strongest adsorption is observed for the S,Obridge configuration, followed by S,O,Obrige,S4F and then Satop modes. In terms of charge transfer, the negative values (Table 3) indicate that electrons were transferred from the Pt surface to the adsorbate. Most charge, i.e. -0.410 e-, was transferred in the S,O,Obridge adsorption mode, where all three atoms of SO2 were bound to the Pt surface. The second highest was in S4F (-0.392 e-), where again the three atoms were bound to the surface, followed by S,Obridge (-0.349 e-) with only the S-O bond aligned to the surface and, finally, Satop (-0.074 e-) where only S was bound to the Pt surface.
We note that on this surface, the adsorption sites for both H2O and SO2 are similar and they will therefore compete directly for adsorption. In one scenario, if we assume that the Pt surface is first covered with H2O on all the adsorption sites, the surface should be saturated with electrons from both the surface and the H2O molecules. If a SO2 molecule were then to approach this water-covered surface, it should easily displace the H2O molecules, as the SO2 can absorb electrons from the surface and has a larger, more favourable adsorption energy, i.e. -2.47 eV for S,Obridgevs -1.68 eV for AtopH2O. Looking at the charge density difference in the S,Obridge configuration, we note that the S-O adsorbed onto the Pt surface has a cumulative charge of 0.79 e-and the O atom directed away from the surface -1.14 e-. This negatively charged O atom would be available for reactions with either surface-bound molecular H2O or dissociated OH- + H+ in the vicinity of the SO2, which could lead to the formation of HSO3-, an intermediary species in the production of hydrogen in the HyS cycle.
3.2.2. Pt (011)
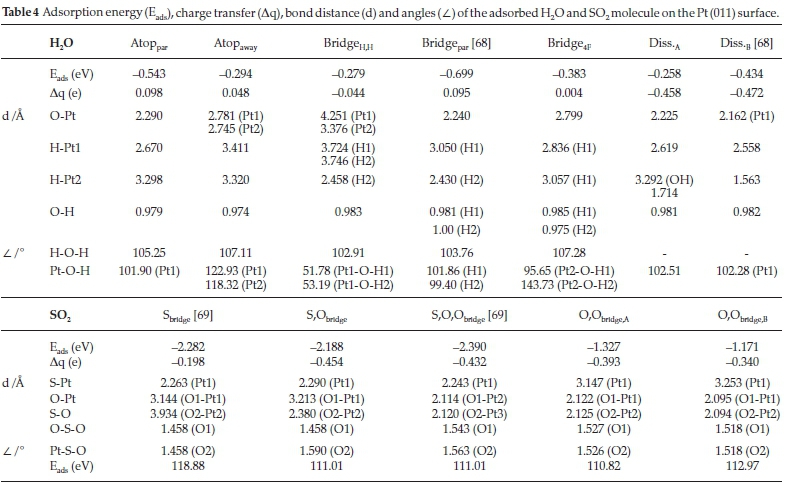
On the (011) surface, five molecularly adsorption modes (Atoppar, Atopaway,Bridgepar and Bridge4Fand two dissociative modes ((011)diss,A and (011)diss,B) were observed for H2O, as shown in Fig. 3. The adsorption energies, charge transfer, bond distances and angles of both H2O and SO2 on the Pt (011) surface are given in Table 4.. In the first adsorption mode, Atoppar, the H2O molecule is parallel to the surface, with the O atom bound atop a Pt atom and the H atoms directed towards the (011) channels. The O-H bond lengths and H-O-H bond angle compare to those in the free molecule, which indicates physisorption. In the second adsorption mode, Atopaway,we found the O atom to be bound between two Pt atoms on the (011) ridge, with the H atoms directed away from the surface. In this adsorption mode, the H-O bond length correlates with the free molecule, although the H-O-H bond angle is larger by ~3 °, which could indicate physisorption. In the third adsorption mode (BridgeH,H), the H atoms were directed towards the surface, forming a bridge across the (011) channel. Surprisingly, the O-H bond length still correlated with the free molecule, even though the H-O-H bond angle was ~1.5 ° smaller, which could also indicate physisorption. In the fourth adsorption mode, Bridgepar, one of the hydrogens of the H2O molecule points in the direction of the ridge on which it is adsorbed, while the other H points towards the neighbouring ridge, as shown in Fig. 3. The O-Pt distances on the (011) surface are somewhat shorter than on the other surfaces, even though the H-O-H angle differs by less than 1 ° from the free molecule. In the fifth adsorption mode, Bridge4F, one O-H is bound across the (011) channel in the 4F position, while the other H atom is directed away from the surface. As expected, the O-H1 bond length in the channel is slightly elongated, while the O-H2 directed away from the surface is similar to the free molecule. The H-O-H bond angle is larger by ~3 °, which again indicates physisorption. Comparing the adsorption energies of the five adsorption modes, it can be seen that the Bridgepar configuration will be favoured, followed byAtoppar, Bridge4F, Atopawayand only then BridgeH,H.
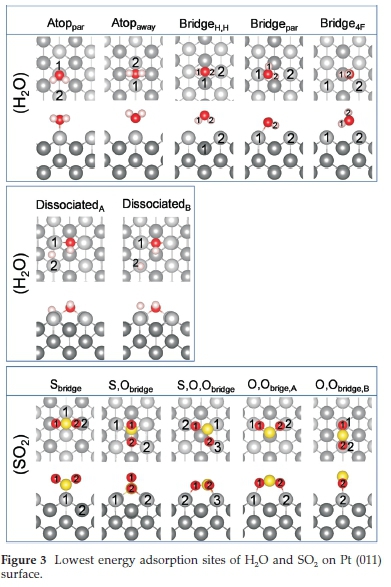
In the dissociated system (Pt (011)diss,A), the OH group lies parallel in the valley of Pt atoms and is bound by its oxygen to the Pt atoms on the neighbouring ridges, following the direction of the valley. Similarly, Shi and Sun106 showed that the dissociated H atom is bound in a bridge position between two Pt atoms on the ridge. In the second dissociated system (Pt (011)diss, B), the OH group is again bound to two Pt atoms on neighbouring ridges, following the direction of the valley. However, the dissociated H atom is bound atop a Pt atom on the ridge. The only difference between Pt (011)diss,A and Pt (011)diss,B is the adsorption energy, which is larger by 0.18 eV for the Pt (011)diss,B adsorption, and thus more favoured.
The relative adsorption energies (Table 4) for the water molecule are smaller than observed on the (001) surface and they follow the trend of Bridgepar > Atoppar > Bridge4F > Atopaway > BridgeHH. However, the adsorption energies of the dissociated water are small and similar to some of the molecularly adsorbed H2O configurations, indicating that there is little incentive for dissociation to occur on the (011) surface. From the charge transfer calculations it can be seen, that, similar to the (001) surface, in molecularly adsorbed H2O, electrons are transferred from the molecule to the Pt surface, which is highest (~ 0.1 e-) for the most favoured configurations Bridgepar and Atoppar. However, it is interesting that the BridgeH,H configuration adsorbs electrons (-0.044 e-) from the Pt surface. With the H atoms directed toward the Pt surface and electrons being donated into the molecule, dissociation could occur, possibly leading to the H2Odiss configurations, although the energetic incentive is low. In the dissociated H2O, charge transfer of between 0.4 and 0.5 e- occurs from the Pt surfaces to the molecule. As expected, the dissociated H atom is electron-depleted (∆q = 1.000 e-) and OH gained nearly an extra 50 % electron density (∆q = -1.458 e-), owing to the adsorption manner of the dissociated H and OH, which are pulled into the (011) framework, thereby favouring electron transfer to and from the Pt surface.
In the adsorption of SO2 on the (011) surface, five possible adsorption modes were observed, i.e. Sbridge, S,Obridge, S,O,Obridge,O,Obridge,Aand O,Obridge,B. In the first adsorption mode, i.e. Sbridge, the SO2 molecule had the same geometry to the Satop configuration on the (001) surface, with the S bound to the Pt surface and the two O atoms directed away from the surface, although here the S atom is located between two ridge Pt atoms. The S-O bond length and O-S-O bond angle was similar to the free SO2 molecule, due to limited interaction between the surface and adsorbate. In the second adsorption mode, i.e. S,Obridge, the S-O bond is parallel to the channel on the (011) surface, with the other O atom directed away from the surface. The S atom is bound across the ridge to two Pt atoms and the O atom to another two Pt atoms of a 4F hollow site. The S-O bond lengths (Table 4) and O-S-O bond angle follow the same trend as for the S,Obridge adsorption mode on the (001) surface where the free S-O bond length is shorter than the bound S-O bond length and the O-S-O bond angle smaller than 119 °, indicating physisorption. In the third adsorption mode, i.e. S,O,Obridge, the SO2 molecule is parallel to the Pt surface with two O atoms bound to two Pt atoms on the (011) ridge, forming an O-O-bridge with Pt diagonally across the (011) channel. Due to the formation of this Pt-O bond, the S-O bond is slightly elongated, while the O-S-O bond angle is smaller than in the free molecule. Similarly, in the fourth adsorption mode, i.e. O,Obridge,A, the SO2 molecule lies parallel to the surface, forming an O-O-bridge with Pt directly across the (011) channel. Again, the S-O bond length is elongated, while the O-S-O bond angle is smaller. In the fifth adsorption mode, O,Obridge,B the molecules also form an O-O-bridge, but with two Pt atoms on the same (011) ridge, with the S atom directed away from the surface. As in the other adsorption modes, the S-O bond lengths are elongated, while the O-S-O bond angle is smaller than in the free molecule.
When we compare the SO2 adsorption energies, it is evident that the most likely adsorption to occur is the S,O,Obridge configuration, followed by Sbridge > S,Obridge > O,Obridge,A> O,Obridge,B. It has been shown in the literature107 for SO2 adsorbed onto Ag (110), that the Sbridge adsorption mode dominates, whereas on the Ni (110) surface, both S,O,Obridge and O,Obrigde,A adsorptions occur.
In terms of charge transfer in the adsorbed SO2 molecules, the trend on the (011) surface is similar to the (001) surface, with Aq the highest where all three atoms (S, O1 and O2) are adsorbed onto the Pt surface, followed by the adsorption of two atoms (S and O or O1 and O2) and then one atom (S). However, on this surface, e.g. in S,Obridge with two adsorbed atoms (S and O2) bound between four Pt atoms, more charge is transferred to SO2, i.e. -0.454 e-, followed by S,O,Obridge (-0.432 e-) with three atoms bound to the surface, next O,Obridge,A (-0.393 e-) with only O-O on the surface, then O,Obridge,B (-0.340 e-) also with O-O on the surface and, finally, Sbridge (-0.198 e-) where only S was bound to the Pt surface.
Comparing the adsorption of H2O and SO2, in terms of the most favourable adsorption energies, we note that the Brigdepar (H2O configuration) and the S,O,Obridge configuration directly compete for adsorption sites. However, in these specific configurations, the H from H2O and the O from SO2 can be directed towards each other for a reaction to occur. A similar result is seen in the second most favourable positions, i.e. Atoppar (H2O configuration) and Sbridge configuration, where their specific binding geometries would allow reaction between H2O and SO2 to occur. If we again assume that the Pt surface is first covered with H2O on all the adsorption sites, an approaching SO2 molecule should be able to displace the H2O as the SO2 then absorbs electrons from the surface. The charge density distributions in the S,O,Obridge and Sbridge configurations are similar to the (001) surface, with a cumulative charge of 0.63 e- and 0.98 e-, respectively, but -1.07 e- and -1.18 e-, respectively, in the O atom directed away from the surface. This negatively charged O atom would be available for reactions with either the molecular H2O or dissociated OH + H in the vicinity as a first step in the HyS process.
3.2.3. Pt (111)
On the (111) surface only one molecular (Atoppar) and one dissociative ((111)diss) adsorption mode for H2O were observed, shown in Fig. 4 with the corresponding adsorption energy, charge transfer, bond distance and angles in Table 5.. In the Atoppar adsorption, the H2O molecule adsorbs parallel to the Pt surface, with one H atom directed towards a surface Pt (Pt1) and the other in the direction of a fcc Pt (Pt2) (Fig. 4). Similarly, Carrasco and co-workers108 showed that the most stable single H2O molecule adsorption was atop the Pt atom and parallel to the surface. In this work, the H-O-H angle correlated with literature at 104.94 and the calculated O-Pt distance was 2.386 A; however, literature reported the O-Pt distance between 2.49 and 2.82 A,108 suggesting that water may bind more strongly to the Pt (111) surface than previously indicated.108 Similar to the (001)diss and (011)diss systems, on the (111) surface the O from the dissociated OH group were bound in a bridge position between two surface Pt atoms. The dissociated hydrogen was in a neighbouring fcc hollow site, which was also reported106 as energetically the most stable adsorption mode for hydrogen on the Pt (111) surface.
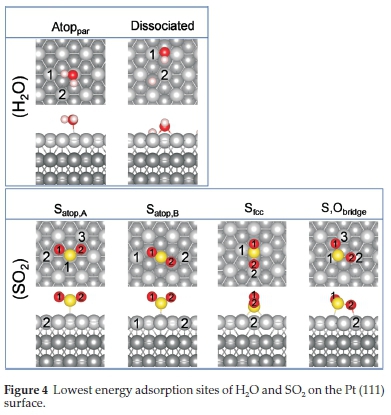
The calculated adsorption energies (Table 5) show that the water molecule does not bind as strongly to the (111) surface as on the (001) and to a lesser extent (011) surfaces, indicating that adsorption and dissociation is favoured on the (001) surface. Literature showed that adsorption energies were dependant on the type of dispersion correction functional used,108 and reported monomer adsorption energies specifically for the Pt (111) surface between -0.24 and -0.40 eV. These values are in fair agreement with our adsorption energy calculated for the (111) surface, but again indicating somewhat stronger binding in this study compared to the literature.108 Comparing our adsorption energies to that of the dissociated water on all the surfaces, we note that generally adsorption is energetically preferred on the (001) surface, both for the molecular and dissociated H2O adsorptions, followed by the (111) and (011) surfaces. On thermodynamic grounds, dissociation should not occur on the (111) surface, where the binding of the dissociated molecule is energetically less favourable than molecular adsorption.
With the adsorption of SO2 on the (111) surface, four possible adsorption modes were observed, i.e. Satop,A,Satop,B,Sfcc and S,Obridge.InSatopA the S atom is bound atop a Pt atom with the O atoms directed (Ptsurface-S-O angle ~18 °) away from the surface. The geometry of the bound SO2 is similar to the free SO2 molecule, indicating physisorption of the molecule. Similarly, in Satop,B, the S is bound atop a Pt atom with the O atoms directed away from the Pt atoms, with a Ptsurface-S-O bond angle of 30 °. This configuration is similar to the Satop adsorption mode on the Pt (001) surface, with S-O bond length and O-S-O bond angle corresponding to the free SO2 molecule, again indicating physisorption of the molecule. In the third adsorption mode, Sfcc, one S-O bond lies in the plane of the surface and the second O atom is directed away from the surface on the fcc binding site. As seen before, the parallel S-O bond is elongated, and the O-S-O angle has decreased due to the binding mode. S,Obridge resembles a configuration between SatopA and Sfcc, where one S-O bond is bound to two atop Pt atoms, with a Ptsurface-S-O2 angle of ~9 °, while the other S-O bond is pointed slightly away from the surface with a Ptsurface-S-O1 angle of ~17 °. The S-O2 bond length is slightly elongated due to the bond with the atop Pt atoms. Lin and co-workers24 identified similar adsorption modes, of which the most likely were Sfcc,SatopB and S,Obridge with adsorption energies ranging between 0.93 and 1.01 eV. When we compare the different adsorption energies, we find that Sfcchas the largest adsorption energy and is therefore the most likely configuration to occur, followed by Satop,b >Satop,B > S,Obridge.
In terms of charge transfer (Table 5) between SO2 and Pt (111), the trend is similar to the (001) and (011) surfaces, with a maximum Aq when all three atoms (S, O1 and O2) are adsorbed onto the Pt surface, although Satop,A with only S adsorbed is an outlier, as this configuration leads to the largest charge transfer (∆q = -0.452 e-). However, the other adsorption modes follow the trend, i.e. three adsorbed atoms (S, O1 and O2) are bound in S,Obridge (-0.370 e-), followed by two adsorbed atoms (S and O2) in Sfcc(-0.240 e-) and finally Satop,B (-0.106 e-) where only S was bound to the Pt surface.
The calculated modes and energies of adsorption of SO2 on the (111) are in fair agreement with the work by Lin and co-workers24 who calculated adsorption energies for the Pt (111) surface ranging between 0.93 and 1.01 eV, depending on adsorption mode and the dispersion correction functional chosen.
If we compare the adsorption sites of both H2O and SO2 on the (111) surface, we note that there is no direct competition for the most favourable adsorption configurations, Atoppar and Sfcc, but there is for the second most favourable configuration, Satop,A.A comparison of the adsorption energies of H2O and SO2 shows a difference of between 1.1 and 1.4 eV, indicating that if a SO2 molecule were to approach a water-covered surface, the H2O could be displaced, as the SO2absorbs electrons from the surface and has a more favourable adsorption energy. Looking at the charge density difference in the Sfccconfiguration, we note that the S-O adsorbed onto the Pt surface has a cumulative charge of 0.91 e- with -1.15 e- in the O atom directed away from the surface. However, in the Satop,A configuration, the charge is distributed equally through the molecule, with a charge transfer of 3.00 e- and -1.72 e- for S and each O atom, respectively. These O atoms may be available for reactions with the H2O molecules.
4. Conclusions
In this study, we have employed density functional theory calculations to predict the behaviour of H2O and SO2 with the Pt (001), (011) and (111) surfaces. Our results show that a H2O molecule will preferentially adsorb dissociatively on the (001) surface, but on both the (011) and (111) surfaces, the H2O molecule adsorbs parallel atop the Pt surface atoms. Charge transfer analysis shows that the molecularly bound H2O provides ~0.1 e- to the Pt surface, whereas in the dissociated case ~0.4 e- transferred from the surface to the molecule.
When SO2 adsorbs onto Pt, was observed that on both the (001) and (111) surfaces, S-O will preferentially adsorb onto the surface with one O atom directed away from the surface. However, on the (011) surface, SO2 lies parallel to the surface with one S-O pair bound across the channel and the other S-O pair bound on the ridge between two Pt atoms. SO2 can act as both a CT-donor or n-acceptor,111 and when the n-acceptor aspect dominates, n bonds are formed between the SO2 and the metal, causing the molecule to adsorb in a parallel orientation on the surface, as seen on the (011) surface. In contrast, when CT-bonding dominates, the molecule adsorbs perpendicular to the surface planes, as shown to occur on the (001) and (111) surfaces. Charge transfer analysis shows that during adsorption, between 0.24 and 0.43 e- is transferred from the surface to the molecule.
Taking into account all the adsorption energy and charge transfer data of both H2O and SO2 on all three Pt surfaces, it can be concluded that generally both molecules compete for the same adsorption sites, where the strong binding of SO2 to the surface sites should enable it to compete effectively to adsorb onto the Pt surfaces at the expense of water adsorption.
Future work will include the consideration of an explicit mixture of H2O and SO2 on the various Pt surfaces, as well as the SO2 oxidation mechanism catalyzed by the Pt metal.
Acknowledgements
We acknowledge the Economic and Social Research Council (ESRC grant No. ES/N013867/1) and the National Research Foundation, South Africa, for a Newton Fund PhD student exchange grant, and the Engineering and Physical Sciences Research Council (EPSRC grants No. EP/K016288/1 and EP/K009567/2) for research funding. This work was performed using the computational facilities of the Advanced Research Computing @ Cardiff (ARCCA) Division; Cardiff University; and the Centre of High-Performance Computing (CHPC) in Cape Town, South Africa. In addition, the calculations were carried out at the Supercomputing Facilities of Cardiff University operated by ARCCA on behalf of the HPC Wales and Supercomputing Wales (SCW) projects. We acknowledge the support of the latter, which is partly funded by the European Regional Development Fund (ERDF) via Welsh Government. We wish to acknowledge the use of the EPSRC-funded National Chemical Database Service hosted by the Royal Society of Chemistry. M.J.U. would like to acknowledge the National Research Foundation (NRF - Grant number 116728) for their financial support and the North-West University for their support and resources. All data created during this research are openly available from Cardiff University's Research Portal at: http://dx.doi.org/10.17035/d.2020.0102392217
ORCID iDs
M.J. Ungerer: orcid.org/0000-0002-9073-1186
D. Santos-Carballal: orcid.org/0000-0002-3199-9588
C.G.C.E. van Sittert: orcid.org/0000-0001-5786-5409
N.H. de Leeuw: orcid.org/0000-0002-8271-0545
References
1 H. Igliñski and M. Babiak, Analysis of the potential of autonomous vehicles in reducing the emissions of greenhouse gases in road transport, Procedia Eng., 2017, 192, 353-358. DOI: 10.1016/j.proeng.2017.06.061 [ Links ]
2 EASA European Aviation Environmental Report 2016, 2019, Vol. 40. [ Links ]
3 Rt Hon Michael Gove Clean Air Strategy Plan, 2019. [ Links ]
4 M. Junginger, E. Hittinger, E. Williams and R. Wiser, Onshore wind energy, in Technological Learning in the Transition to a Low-Carbon Energy System, Elsevier, 2020, pp. 87-102. [ Links ]
5 P. Varela-Vázquez, M. del C. Sánchez-Carreira and Ó. Rodil-Marzábal, A novel systemic approach for analysing offshore wind energy implementation, J. Clean. Prod., 2019, 212, 1310-1318. DOI: 10.1016/j.jclepro.2018.12.079 [ Links ]
6 T. Sarver, A. Al-Qaraghuli and L.L. Kazmerski, A comprehensive review of the impact of dust on the use of solar energy: history, investigations, results, literature, and mitigation approaches, Renew. Sustain. Energy Rev., 2013, 22, 698-733. DOI: 10.1016/j.rser.2012.12.065 [ Links ]
7 J. Gong, C. Li and M.R. Wasielewski, Advances in solar energy conversion, Chem. Soc. Rev., 2019,48, 1862-1864. DOI: 10.1039/C9CS90020A [ Links ]
8 A.J. Carrillo, J. González-Aguilar, M. Romero and J.M. Coronado, Solar energy on demand: a review on high temperature thermo-chemical heat storage systems and materials, Chem. Rev., 2019, 119, 4777-4816. DOI: 10.1021/acs.chemrev.8b00315 [ Links ]
9 G. Falchetta, D.E.H.J. Gernaat, J. Hunt and S. Sterl, Hydropower dependency and climate change in sub-Saharan Africa: a nexus framework and evidence-based review, J. Clean. Prod., 2019, 231, 1399-1417. DOI: 10.1016/j.jclepro.2019.05.263 [ Links ]
10 S. Sharma, J. Waldman, S. Afshari and B. Fekete, Status, trends and significance of American hydropower in the changing energy landscape, Renew. Sustain. Energy Rev., 2019,101, 112-122. DOI: 10.1016/j.rser.2018.10.028 [ Links ]
11 J. Jurasz, F.A. Canales, A. Kies, M. Guezgouz and A. Beluco, A review on the complementarity of renewable energy sources: concept, metrics, application and future research directions, Sol. Energy, 2020, 195, 703-724. DOI: 10.1016/j.solener.2019.11.087 [ Links ]
12 M. Edwin, M.S. Nair and S. Joseph Sekhar, A comprehensive review for power production and economic feasibility on hybrid energy systems for remote communities, Int. J. Ambient Energy, 2020, 1-13. DOI: 10.1080/01430750.2020.1712252 [ Links ]
13 E. van der Roest, L. Snip, T. Fens and A. van Wijk, Introducing Power-to-H3: combining renewable electricity with heat, water and hydrogen production and storage in a neighbourhood, Appl. Energy, 2020, 257, 114024. DOI: 10.1016/j.apenergy.2019.114024 [ Links ]
14 F. de Bruijn, The current status of fuel cell technology for mobile and stationary applications, Green Chem., 2005, 7, 132-150. DOI: 10.1039/b415317k [ Links ]
15 P. Nikolaidis and A. Poullikkas, A comparative overview of hydrogen production processes, Renew. Sustain. Energy Rev., 2017, 67, 597-611. DOI: 10.1016/j.rser.2016.09.044 [ Links ]
16 V. Singh and D. Das, Chapter3-Potential of hydrogen production from biomass, in Science and Engineering of Hydrogen-Based Energy Technologies, Miranda, P.E.V. de, Ed., Academic Press, 2018, pp. 123-164. ISBN 978-0-12-814251-6 [ Links ]
17 S.T. Revankar, Chapter 4 - Nuclear hydrogen production, in Storage and Hybridization of Nuclear Energy, Bindra,, H., Revankar, S., Eds., Academic Press, 2019, pp. 49-117. ISBN 978-0-12-813975-2 [ Links ]
18 S.Z. Ahammad and T.R. Sreekrishnan, Energy from wastewater treatment, in Bioremediation and Bioeconomy, Elsevier, 2016, pp. 523-536. [ Links ]
19 L. Xue, P. Zhang, S. Chen and L. Wang, Pt-based bimetallic catalysts for SO2-depolarized electrolysis reaction in the hybrid sulfur process, Int. J. Hydrogen Energy, 2014, 39, 14196-14203. DOI: 10.1016/j.ijhydene.2014.02.128 [ Links ]
20 J.A. O'Brien, J.T. Hinkley, S.W. Donne and S.E. Lindquist, The electro-chemical oxidation of aqueous sulfur dioxide: a critical review of work with respect to the hybrid sulfur cycle, Electrochim. Acta, 2010, 55, 573-591. DOI: 10.1016/j.electacta.2009.09.067 [ Links ]
21 A.J. Appleby and B. Pinchon, Electrochemical aspects of the H2SO4SO2 thermoelectrochemical cycle for hydrogen production, Int. J. Hydrogen Energy, 1980, 5, 253-267. DOI: 10.1016/0360-3199(80)90070-1 [ Links ]
22 P.W.T. Lu and R.L. Ammon, An investigation of electrode materials for the anodic oxidation of sulfur dioxide in concentrated sulfuric acid, J. Electrochem. Soc., 1980, 127, 2610. DOI: 10.1149/1.2129530 [ Links ]
23 H.R. Colón-Mercado and D.T. Hobbs, Catalyst evaluation for a sulfur dioxide-depolarized electrolyzer, Electrochem. Commun., 2007, 9, 2649-2653. DOI: 10.1016/j.elecom.2007.08.015 [ Links ]
24 X. Lin, K.C. Hass, W.F. Schneider and B.L. Trout, Chemistry of sulfur oxides on transition metals i: configurations, energetics, orbital analyses, and surface coverage effects of SO 2 on Pt(111), J. Phys. Chem. B, 2002,106, 12575-12583. DOI: 10.1021/jp026128f [ Links ]
25 G.A. Somorjai, On the mechanism of sulfur poisoning of platinum catalysts, J. Catal, 1972, 27, 453-456. DOI: 10.1016/0021-9517(72)90183-2 [ Links ]
26 N.S. Nasri, J.M. Jones, VA. Dupont and A. Williams, A comparative study of sulfur poisoning and regeneration of precious-metal catalysts, Energy & Fuels, 1998,12, 1130-1134. DOI: 10.1021/ef980104j [ Links ]
27 M. Polcik, L. Wilde, J. Haase, B. Brena, D. Cocco, G. Comelli and G. Paolucci, Adsorption and temperature-dependent decomposition of SO2 on Cu(100) and Cu(111): a fast and high-resolution core-level spectroscopy study, Phys. Rev. B, 1996, 53, 13720-13724. DOI: 10.1103/PhysRevB.53.13720 [ Links ]
28 M. Polèik, L. Wilde and J. Haase, SO2-induced surface reconstruction of Cu(111): an x-ray-absorption fine-structure study, Phys. Rev. B, 1998, 57, 1868-1874. DOI: 10.1103/PhysRevB.57.1868 [ Links ]
29 M.S. Wilburn and W.S. Epling, SO2 adsorption and desorption characteristics of Pd and Pt catalysts: precious metal crystallite size dependence, Appl. Catal. A Gen., 2017, 534, 85-93. DOI: 10.1016/j.apcata.2017.01.015 [ Links ]
30 J.A. Rodriguez, J.M. Ricart, A. Clotet and F. Illas, Density functional studies on the adsorption and decomposition of SO2 on Cu(100), J. Chem. Phys., 2001,115, 454-465. DOI: 10.1063/1.1377884 [ Links ]
31 T. Yokoyama, S. Terada, S. Yagi, A. Imanishi, S. Takenaka, Y. Kitajima and T. Ohta, Surface structures and electronic properties of SO2 adsorbed on Ni(111) and Ni(100) studied by S K-edge X-ray absorption fine structure spectroscopy, Surf. Sci., 1995, 324, 25-34. DOI: 10.1016/0039-6028(94)00692-X [ Links ]
32 S. Terada, A. Imanishi, T. Yokoyama, S. Takenaka, Y. Kitajima and T. Ohta, Surface structure of SO2 adsorbed on Ni(110) studied by S K-edge X-ray absorption fine structure spectroscopy, Surf. Sci., 1995, 336, 55-62. DOI: 10.1016/0039-6028(95)00514-5 [ Links ]
33 P. Zebisch, M. Weinelt and H.-P. Steinrück, Sulphur dioxide adsorption on the Ni(110) surface, Surf. Sci., 1993, 295, 295-305. DOI: 10.1016/0039-6028(93)90276-P [ Links ]
34 J. Ahner, A. Effendy, K. Vajen and H.-W. Wassmuth, Chemisorption and multilayer adsorption of SO2 on Ag(111) and Ag(110), Vacuum, 1990,41, 98-101. DOI: 10.1016/0042-207X(90)90285-7 [ Links ]
35 J.L. Solomon, R.J. Madix, W. Wurth and J. Stohr, NEXAFS and EELS study of the orientation of sulfur dioxide on silver(110), J. Phys. Chem., 1991, 95, 3687-3691. DOI: 10.1021/j100162a046 [ Links ]
36 R.C. Ku and P. Wynblatt, SO2 adsorption on Rh(110) and Pt(110) surfaces, Appl. Surf. Sci., 1981, 8, 250-259. DOI: 10.1016/0378-5963(81)90120-3 [ Links ]
37 J.A. Rodriguez, T. Jirsak and S. Chaturvedi, Reaction of S2 and SO2 with Pd/Rh(111) surfaces: effects of metal-metal bonding on sulfur poisoning, J. Chem. Phys., 1999,110, 3138-3147. DOI: 10.1063/1.477910 [ Links ]
38 S. Terada, M. Sakano, Y. Kitajima, T. Yokoyama and T. Ohta, Adsorption of SO2 on Pd(100) studied by S K-Edge XAFS, Le J. Phys. IV, 1997, 7, C2-703-C2-704. DOI: 10.1051/jp4:1997211 [ Links ]
39 M.L. Burke and R.J. Madix, Hydrogen on Pd(100)-S: the effect of sulfur on precursor mediated adsorption and desorption, Surf. Sci., 1990, 237, 1-19. DOI: 10.1016/0039-6028(90)90515-A [ Links ]
40 J.M. Saleh, Interaction of sulphur compounds with palladium, Trans. Faraday Soc., 1970, 66, 242. DOI: 10.1039/tf9706600242 [ Links ]
41 M.L. Burke and R.J. Madix, SO2 structure and reactivity on clean and sulfur modified Pd(100), Surf. Sci., 1988,194, 223-244. DOI: 10.1016/0039-6028(94)91257-2 [ Links ]
42 S. Astegger and E. Bechtold, Adsorption of sulfur dioxide and the interaction of coadsorbed oxygen and sulfur on Pt(111), Surf. Sci., 1982,122, 491-504. DOI: 10.1016/0039-6028(82)90098-X [ Links ]
43 U. Köhler and H.-W. Wassmuth, SO2 adsorption and desorption kinetics on Pt(111), Surf. Sci., 1983,126, 448-454. DOI: 10.1016/0039-6028(83)90742-2 [ Links ]
44 Y.-M. Sun, D. Sloan, D.J. Alberas, M. Kovar, Z.-J. Sun and J.M. White, SO2 adsorption on Pt(111): HREELS, XPS and UPS study, Surf. Sci., 1994, 319, 34-44. DOI: 10.1016/0039-6028(94)90567-3 [ Links ]
45 M. Polèik, L. Wilde, J. Haase, B. Brena, G. Comelli and G. Paolucci, High-resolution XPS and NEXAFS study of SO2 adsorption on Pt(111): two surface SO2 species, Surf. Sci., 1997, 381, L568-L572. DOI: 10.1016/S0039-6028(97)00060-5 [ Links ]
46 X. Lin, W.F. Schneider and B.L. Trout, Chemistry of sulfur oxides on transition metals. II. Thermodynamics of sulfur oxides on platinum-(111), J. Phys. Chem. B, 2004,108, 250-264. DOI: 10.1021/jp035306h [ Links ]
47 C.-H. Chen, A. Halford, M. Walker, C. Brennan, S.C.S. Lai, D.J. Fermin, P.R. Unwin and P. Rodriguez, Electrochemical characterization and regeneration of sulfur poisoned Pt catalysts in aqueous media, J. Electroanal. Chem., 2018, 816, 138-148. DOI: 10.1016/j.jelechem.2018.03.015 [ Links ]
48 J. Oudar, Sulfur adsorption and poisoning of metallic catalysts, Catal. Rev., 1980, 22, 171-195. DOI: 10.1080/03602458008066533 [ Links ]
49 T.J. Truex, Interaction of sulfur with automotive catalysts and the impact on vehicle emissions - A review, SAE Tech. Pap., 1999, 108, 1192-1206. DOI: 10.4271/1999-01-1543 [ Links ]
50 S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C. Liu, A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: nanostructure, activity, mechanism and carbon support in PEM fuel cells, J. Mater. Chem. A, 2017, 5, 1808-1825. DOI: 10.1039/C6TA08580F [ Links ]
51 H. Luo, S. Park, H.Y.H. Chan and M.J. Weaver, Surface Oxidation of platinum-group transition metals in ambient gaseous environments: role of electrochemical versus chemical pathways, J. Phys. Chem. B, 2000, 104, 8250-8258. DOI: 10.1021/jp001289+ [ Links ]
52 T. Pradeep, Anshup noble metal nanoparticles for water purification: a critical review, Thin Solid Films, 2009, 517, 6441-6478. DOI: 10.1016/j.tsf.2009.03.195 [ Links ]
53 R.A. Goyer, Toxic and essential metal interactions, Annu. Rev. Nutr., 1997,17, 37-50. DOI: 10.1146/annurev.nutr.17.1.37 [ Links ]
54 Hodgson and S. Haq, Water adsorption and the wetting of metal surfaces, Surf. Sci. Rep., 2009, 64, 381-451. DOI: 10.1016/j.surfrep.2009.07.001 [ Links ]
55 L.Q. Guo, X.M. Zhao, Y. Bai and L.J. Qiao, Water adsorption behavior on metal surfaces and its influence on surface potential studied by in situ SPM, Appl. Surf. Sci., 2012, 258, 9087-9091. DOI: 10.1016/j.apsusc.2012.06.003 [ Links ]
56 J. Carrasco, A. Hodgson and A. Michaelides, A molecular perspective of water at metal interfaces, Nat. Mater., 2012, 11, 667-674. DOI: 10.1038/nmat3354 [ Links ]
57 L. Bellarosa, R. García-Muelas, G. Revilla-López and N. López, Diversity at the water-metal interface: metal, water thickness, and confinement effects, ACS Cent. Sci., 2016, 2, 109-116. DOI: 10.1021/acscentsci.5b00349 [ Links ]
58 S. Meng, E.G. Wang and S. Gao, Water adsorption on metal surfaces: a general picture from density functional theory studies, Phys. Rev. B, 2004, 69, 195404. DOI: 10.1103/PhysRevB.69.195404 [ Links ]
59 S. Kenmoe and P. Ulrich Biedermann, Water aggregation and dissociation on the ZnO(1010) surface, Phys. Chem. Chem. Phys., 2017, 19, 1466-1486. DOI: 10.1039/c6cp07516a [ Links ]
60 G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B, 1993, 47, 558-561. DOI: 10.1103/PhysRevB.47.558 [ Links ]
61 G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metalamorphous-semiconductor transition in germanium, Phys. Rev. B, 1994, 49, 14251-14269. DOI: 10.1103/PhysRevB.49.14251 [ Links ]
62 G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B, 1996, 54, 11169-11186. DOI: 10.1103/PhysRevB.54.11169 [ Links ]
63 G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15-50. DOI: 10.1016/0927-0256(96)00008-0 [ Links ]
64 S. Posada-Pérez, D. Santos-Carballal, U. Terranova, A. Roldan, F. Illas and N.H. de Leeuw, CO2 interaction with violarite (FeNi2S4) surfaces: a dispersion-corrected DFT study, Phys. Chem. Chem. Phys., 2018, 20, 20439-20446. DOI: 10.1039/C8CP03430C [ Links ]
65 N.Y. Dzade, A. Roldan and N.H. de Leeuw, Activation and dissociation of CO 2 on the (001), (011), and (111) surfaces of mackinawite (FeS): a dispersion-corrected DFT study, J. Chem. Phys., 2015, 143, 094703. DOI: 10.1063/1.4929470 [ Links ]
66 A.K. Mishra, A. Roldan and N.H. de Leeuw, CuO Surfaces and CO2 activation: a dispersion-corrected DFT+ U study, J. Phys. Chem. C, 2016,120, 2198-2214. DOI: 10.1021/acs.jpcc.5b10431 [ Links ]
67 D. Santos-Carballal, A. Roldan, N.Y. Dzade and N.H. de Leeuw, Reactivity of CO2 on the surfaces of magnetite (Fe3O4), greigite (Fe3S4) and mackinawite (FeS), Philos. Trans. R. Soc. A Math. Phys. Eng. Sci., 2018, 376, 20170065. DOI: 10.1098/rsta.2017.0065 [ Links ]
68 M.J. Ungerer, D. Santos-Carballal, A. Cadi-Essadek, C.G.C.E. van Sittert and N.H. de Leeuw, Interaction of H2O with the platinum Pt (001), (011) and (111) surfaces: a density functional theory study with long-range dispersion corrections, J. Phys. Chem. C, 2019, 132, 27465-27476. DOI: 10.1021/acs.jpcc.9b06136 [ Links ]
69 M.J. Ungerer, D. Santos-Carballal, A. Cadi-Essadek, C.G.C.E. van Sittert and N.H. de Leeuw, Interaction of SO2 with the platinum (001), (011) and (111) surfaces: a DFT study, Catalysts, 2020,10, 558. [ Links ]
70 P.E. Blöchl, Projector augmented-wave method, Phys. Rev. B, 1994,50, 17953-17979. DOI: 10.1103/PhysRevB.50.17953 [ Links ]
71 G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B, 1999,59,1758-1775. DOI: 10.1103/PhysRevB.59.1758 [ Links ]
72 J.P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865-3868. DOI: 10.1103/PhysRevLett.77.3865 [ Links ]
73 J.P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple (Errata), Phys. Rev. Lett., 1996, 77, 3865-3868. DOI: 10.1103/PhysRevLett.77.3865 [ Links ]
74 S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456-1465. DOI: 10.1002/jcc.21759 [ Links ]
75 M. Methfessel and A.T. Paxton, High-precision sampling for Brillouin-zone integration in metals, Phys. Rev. B, 1989,40,3616-3621. DOI: 10.1103/PhysRevB.40.3616 [ Links ]
76 P.E. Blöchl, O. Jepsen and O.K. Andersen, Improved tetrahedron method for Brillouin-zone integrations, Phys. Rev. B, 1994, 49, 16223-16233. DOI: 10.1103/PhysRevB.49.16223 [ Links ]
77 G. Corbel, M. Topic, A. Gibaud and C.I. Lang, Selective dry oxidation of the ordered Pt-11.1 at.% v alloy surface evidenced by in situ temperature-controlled X-ray diffraction, J. Alloys Compd., 2011, 509, 6532-6538. DOI: 10.1016/j.jallcom.2011.03.079 [ Links ]
78 H.J. Monkhorst and J.D. Pack, Special points for Brillouin-zon integrations, Phys. Rev. B, 1976,13, 5188-5192. DOI: 10.1103/PhysRevB.16.1748 [ Links ]
79 J.W. Arblaster, Crystallographic properties of platinum, Platin. Met. Rev., 1997,41, 12-21. http://www.technology.matthey.com/article/41/1/12-21/ [ Links ]
80 J.W. Arblaster, Crystallographic properties of platinum (Errata), Platin. Met. Rev., 2006, 50, 118-119. DOI: 10.1595/147106706X129088 [ Links ]
81 G.W. Watson, E.T. Kelsey, N.H. de Leeuw, D.J. Harris and S.C. Parker, Atomistic simulation of dislocations, surfaces and interfaces in MgO, J. Chem. Soc. Faraday Trans., 1996, 92, 433. DOI: 10.1039/ft9969200433 [ Links ]
82 N.D. Lang and W. Kohn, Theory of metal surfaces: work function, Phys. Rev. B, 1971, 3, 1215-1223. DOI: 10.1103/PhysRevB.3.1215 [ Links ]
83 G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354-360. DOI: 10.1016/j.commatsci.2005.04.010 [ Links ]
84 E. Sanville, S.D. Kenny, R. Smith and G. Henkelman, Improved grid-based algorithm for Bader charge allocation, J. Comput. Chem., 2007, 28, 899-908. DOI: 10.1002/jcc.20575 [ Links ]
85 W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys. Condens. Matter, 2009,21,084204. DOI: 10.1088/0953-8984/21/8/084204 [ Links ]
86 M. Yu and D.R. Trinkle, Accurate and efficient algorithm for Bader charge integration, J. Chem. Phys., 2011,134, 1-8. DOI: 10.1063/1.3553716 [ Links ]
87 H.L. Skriver and N.M. Rosengaard, Surface energy and work function of elemental metals, Phys. Rev. B, 1992, 46, 7157-7168. DOI: 10.1103/PhysRevB.46.7157 [ Links ]
88 N.H. De Leeuw and C.J. Nelson, A computer modeling study of perfect and defective silver (111) surfaces, J. Phys. Chem. B ,2003,107,3528-3534. DOI: 10.1021/jp027001t [ Links ]
89 N.E. Singh-Miller and N. Marzari, Surface energies, work functions, and surface relaxations of low-index metallic surfaces from first principles, Phys. Rev. B - Condens. Matter Mater. Phys., 2009, 80. DOI: 10.1103/PhysRevB.80.235407 [ Links ]
90 Z. Jian-Min, M. Fei and X. Ke-Wei, Calculation of the surface energy of fcc metals with modified embedded-atom method, Chinese Phys., 2004,13, 1082-1090. DOI: 10.1088/1009-1963/13/7/020 [ Links ]
91 B.J. Keene, Review of data for the surface tension of pure metals, int. Mater. Rev., 1993, 38, 157-192. DOI: 10.1179/imr.1993.38.4.157 [ Links ]
92 W.R.Tyson and WA. Miller, Surface free energies of solid metals: estimation from liquid surface tension measurements, Surf. Sci., 1977,62, 267-276. DOI: 10.1016/0039-6028(77)90442-3 [ Links ]
93 H. Xin and S. Linic, Communications: Exceptions to the d-band model of chemisorption on metal surfaces: the dominant role of repulsion between adsorbate states and metal d-states, J. Chem. Phys., 2010,132. DOI: 10.1063/1.3437609 [ Links ]
94 Caglar, A.C. Kizilkaya, J.W. Niemantsverdriet and C.J. Weststrate, Application of work function measurements in the study of surface catalyzed reactions on Rh(1 0 0), Catal. Struct. React. 2018, 4, 1-11. DOI: 10.1080/2055074X.2018.1434986 [ Links ]
95 J.K. Norskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Density functional theory in surface chemistry and catalysis, Proc. Natl. Acad. Sci., 2011,108, 937-943. DOI: 10.1073/pnas.1006652108 [ Links ]
96 A.R. Hoy and P.R. Bunker, A precise solution of the rotation bending Schrödinger equation for a triatomic molecule with application to the water molecule, J. Mol. Spectrosc., 1979, 74, 1-8. DOI: 10.1016/0022-2852(79)90019-5 [ Links ]
97 R.H. Tromp, P. Postorino, G.W. Neilson, M.A. Ricci and A.K. Soper, Neutron diffraction studies of H2O/D2O at supercritical temperatures. A direct determination of g HH (r), g OH (r), and g OO (r), J. Chem. Phys., 1994,101, 6210-6215. DOI: 10.1063/1.468403 [ Links ]
98 A.H. Clark and B. Beagley, Electron-diffraction investigations of gaseous sulphur dioxide and trioxide, Trans. Faraday Soc., 1971,67,2216. DOI: 10.1039/tf9716702216 [ Links ]
99 P.A. Thiel and T.E. Madey, The interaction of water with solid surfaces: fundamental aspects, Surf. Sci. Rep., 1987, 7, 211-385. DOI: 10.1016/0167-5729(87)90001-X [ Links ]
100 M. Henderson, The interaction of water with solid surfaces: fundamental aspects revisited, Surf. Sci. Rep., 2002, 46, 1-308. DOI: 10.1016/S0167-5729(01)00020-6 [ Links ]
101 P.J. Feibelman, Partial dissociation of water on Ru(0001), Science, 2002, 295, 99-102. DOI: 10.1126/science.1065483 [ Links ]
102 R. Ludwig, How does water bind to metal surfaces: hydrogen atoms up or hydrogen atoms down? Angew. Chemie Int. Edn., 2003, 42, 3458-3460. DOI: 10.1002/anie.200301658 [ Links ]
103 G.J. Kubas, Diagnostic Features of transition-metal-S02 coordination geometries, Inorg. Chem., 1979,18, 182. [ Links ]
104 W.S. Benedict, N. Gailar and E.K. Plyler, Rotation-vibration spectra of deuterated water vapor, J. Chem. Phys., 1956, 24, 1139-1165. DOI: 10.1063/1.1742731 [ Links ]
105 T. Jirsak, J.A. Rodriguez, S. Chaturvedi and J. Hrbek, Chemistry of SO2 on Ru(001):formation ofSO3 and SO4, Surf. Sci., 1998,418,8-21. DOI: 10.1016/S0039-6028(98)00652-9 [ Links ]
106 Q. Shi and R. Sun, Adsorption manners of hydrogen on Pt(10 0), (11 0) and (111) surfaces at high coverage, Comput. Theor. Chem., 2017, 1106, 43-49. DOI: 10.1016/j.comptc.2017.02.024 [ Links ]
107 J. Haase, Structural studies of SO2 adsorption on metal surfaces, J. Phys. Condens. Matter, 1997, 9, 3647-3670. DOI: 10.1088/0953-8984/9/18/006 [ Links ]
108 J. Carrasco, J. Klimes and A. Michaelides, The role of van der Waals forces in water adsorption on metals, J. Chem. Phys., 2013,138. DOI: 10.1063/1.4773901 [ Links ]
109 Michaelides, V.A. Ranea, P.L. de Andres and D.A. King, General model for water monomer adsorption on close-packed transition and noble metal surfaces, Phys. Rev. Lett., 2003, 90, 216102. DOI: 10.1103/PhysRevLett.90.216102 [ Links ]
110 M. Happel, N. Luckas, F. Viñes, M. Sobota, M. Laurin and J. Libuda, SO2 Adsorption on Pt(111) and oxygen precovered Pt(111): a combined infrared reflection absorption spectroscopy and density functional study, J. Phys. Chem. C, 2011, 115, 479-491. DOI: 10.1021/jp107171t [ Links ]
111 R.R. Ryan, G.J. Kubas, D.C. Moody and P.G. Eller, Structure and bonding of transition metal-sulfur dioxide complexes, in Structural Bonding, 1981, Vol. 46, pp. 47-100. [ Links ]
Received 14 April 2020
Revised 10 August 2020
Accepted 7 October 2020
* To whom correspondence should be addressed. E-mail: C.G.C.E.v.S., cornle.vanslttert@nwu.ac.za / N.H.d.L., n.h.deleeuw@uu.nl


{kind=link}
{kind=link}
{kind=link}
{kind=link}