










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.74 Durban 2021
http://dx.doi.org/10.17159/0379-4350/2021/v74a9
RESEARCH ARTICLE
Catalytic Hydrogenation of Sorbic Acid using Pyrazolyl Palladium(II) and Nickel(II) Complexes as Precatalysts
Oluwasegun E. OlaoyeI; Olayinka OyetunjiI; Banothile C.E. MakhubelaII; Apollinaire MuyanezaI, IV; Gopendra KumarI; James DarkwaII, III, *
IDepartment of Chemistry, University of Botswana, Private Bag UB 00704, Gaborone, Botswana
IIDepartment of Chemical Sciences, University of Johannesburg, Kingsway Campus, Auckland Park, 2006, South Africa
IIIBotswana Institute for Technology Research and Innovation, Machel Drive, Gaborone, Botswana
IVCREDERE Associates, LLC, 776 Main Street, Westbrook, ME 04092, USA
ABSTRACT
We have prepared several pyrazolyl palladium and nickel complexes ([(L1)PdCl2](1), [(L2) PdCl2](2), [(L3) PdCl2](3), [(L1) NiBr2](4), [(L2) NiBr2](5) and [(L3) NiBr2](6)) by reacting 3,5-dimethyMH-pyrazole (L1), 3,5-di-ferf-butyl-1ZÏ-pyrazole (L2) and 5-ferrocenyl-1Zf-pyrazole(L3) with [PdCl2(NCMe)2] or [NiBr2(DME)] to afford mononuclear palladium and nickel complexes, respectively. These complexes were then investigated as pre-catalysts in the hydrogenation of 2,4-hexadienoic acid (sorbic acid). The active catalysts from these complexes demonstrate significant activities under mild experimental conditions. Additionally, the active catalysts show that the hydrogenation of sorbic acid proceeds in a sequential manner, where the less hindered C=C bond (4-hexenoic acid) is preferentially reduced over the more hindered C=C bond (2-hexenoic acid).
Keywords: Pyrazolyl catalysts, sorbic acid, hydrogenation, selectivity.
1. Introduction
Hydrogenation of a,ß-unsaturated compounds has been widely employed in the vitamins, fragrances, pharmaceuticals, petrochemicals, agrochemicals, and cosmetics industries.1 One of the extensively used transition metal catalysts in these hydrogenation reactions is chlorotris(triphenylphosphine)rhodium(I), [RhCl(PPh3)3].2,3 The catalyst, RhCl(PPh3)3, catalyzes the chemo-specific hydrogenation of C=C bonds in the presence of other easily reduced groups, like nitro (NO2) or carbonyl (CHO), as well as terminal alkenes even when the substrate has internal alkenes.2,3 Other transition metals complexes have been extensively studied as heterogeneous and homogeneous catalysts in the catalytic hydrogenation of olefins and a,ß-unsaturated compounds. Among these metal complexes are ruthenium,4,5 rhodium,6 iridium,7 and platinum.8 However, nickel9 and palladium10 complexes have in recent times gained considerable attention as efficient catalysts in hydrogenation reactions. Shevlin et al. described the first homogeneous nickel-catalyzed asymmetric hydrogenation of a,ß-unsaturated esters, using molecular hydrogen, that gave high yield and high enantio-selective products.11 Apart from the good reactivity and selectivity of the nickel catalysts, ligand manipulation makes them attractive for homogeneous catalysts. Nickel catalysts are cheap and cost-effective.
On the other hand, palladium catalysts are more expensive but exhibit superior catalytic and selectivity properties in the hydrogenation of unsaturated compounds.12 For example, P^C^P palladium pincer complexes are highly active catalysts for the chemo-selective transfer hydrogenation of a,ß-unsaturated ketones.13 These highly reactive and selective palladium pincer complexes afforded saturated ketones from a-enones.13 Similarly, Bacci et al. reported hydrazinic-phosphine(P^N)palladium(II) complexes as efficient catalysts for C=C bonds hydrogenation under mild experimental condition.14
However, because phosphines are sensitive to air and moisture,15 palladium complexes with nitrogen-donor ligands are emerging as an alternative to phosphorus-donor palladium complexes as hydrogenation catalysts. For example, {bis(aryl-imino)acenaphthene}-palladium(0) complexes are known to be efficient and highly chemo-selective in the hydrogenation of C=C bonds of a,ß-unsaturated aldehydes.16 But despite numerous nitrogen-donor nickel(II) and palladium(II) applications in catalysis, little work has been reported on their catalytic properties in the hydrogenation of a,ß-unsaturated compounds.
In this study, we report on pyrazolyl nickel(II) and palladium(II) complexes as catalysts for the hydrogenation of 2,4-hexanoic acid (sorbic acid), which is an a,ß-unsaturated acid. This study forms part of a bigger project on partial hydrogena-tion of biofuels from triglycerides.
2. Experimental
2.1. General Information
Standard Schlenk and vacuum line techniques were used to handle all air and moisture sensitive compounds. All chemicals and gases were procured from the sources indicated for each one of them and include their purity: Gases - argon and hydrogen (>99 % purity) from Afrox (South Africa); solvents and reagents from Sigma Aldrich - Ethylformate (97 %), acetic anhydride (99 %), ferrocene (98 %hydrazine monohydrate (98 %), hydra-zine dihydrochloride (98 %), (ethyleneglycoldimethylether) nickel(II) bromide (98 %), formic acid (95 %) and 3,5-dimethyl-1H-pyrazole (L1) (99 %).
Literature procedures were used to prepare the following starting materials: 3,5-di-tert-butyl-1H-pyrazole (L2)17, 3-ferro-cenyl-1H-pyrazole (L3)18and [PdCl2(NCMe)2]19 as well as dibromo{bis-3,5-dimethyl-1H-pyrazole}nickel(II) (4)and dibromo{bis-3,5-tert-butyl-1H-pyrazole}nickel(II) (5)20(a)with L1 and L2, respectively.
NMR spectra were recorded in CDCl3 as solvent using Bruker 400 Ultra-shield MHz NMR spectrometer at 400 MHz for the 1H spectra and 100 MHz for the 13C{1H} spectra. Infrared spectra were recorded on a Perkin Elmer FT-IR Spectrum BX II fitted with an ATR probe. Melting points were determined using Gallenkamp Digital Melting-point Apparatus 5A 6797, while elemental analysis data were collected on a Thermos Scientific FLASH 2000 CHNS-O Analyser. Mass spectra were similarly collected on a Waters API Quattro Micro Triple Quadrupole electrospray ionization mass spectrometer.
All hydrogenation reactions were carried out in PPV-CTR01-CE (Eyela, Japan) high-pressure autoclave reactor with a stirring pact, heating and cooling systems.20(b) The course of hydrogena-tion reactions involving palladium catalysts were followed by 1H NMR spectroscopy, using dioxane as an internal standard, which was used to determine percentage conversions. The conversions were determined using the diagnostic peaks, following the integrations of the products from the hydrogenation reaction compared to the integration of dioxane.
2.2. Syntheses of bis(Pyrazole)palladium(II) and Nickel(II) Complexes
2.2.1. Synthesis of Dichloro{bis-3,5-dimethyl-1H-pyrazole} palladium(II) (1)
L1 (74 mg, 0.771 mmol) and [PdCl2(NCMe)2] (100 mg, 0.386 mmol) were dissolved in CH2Cl2 (20 mL), and then stirred continuously at room temperature for 24 h to produce an orange solution. This was followed by in vacuo removal of the solvent to produce compound 1 as an orange solid. Yield: 150 mg (84 %); melting point: 250-252 °C (decomposes without melting). 1H NMR (400 MHz, CDCl3): 1H NMR (CDCl3): á(ppm) 1.89 (s, 2x3H, 2xCH3); 2.65 (s, 2x3H, 2xCH3); 5.67 (s, 1H, 4-H pz); 11.82 (s, 1H, N-H pz) (Fig. SI-8). 13C{1H} NMR (100 MHz, CDCl3) (ppm): 151.6 (Cd-C); 143.0 (Ce-C); 105.6 (Cc-CH); 14.9 (Cb-CH3); 10.3 (Ca-CH3) (Fig. SI-9). Elemental analysis; Anal. calcd. for C10H16Cl2N4Pd: C, 32.50 %; H, 4.36 %; N, 15.16 %. Found: C, 32.92 %; H, 4.34 %; N, 15.06 %.
2.2.2. Synthesis of Dichloro{bis-3,5-tert-buytl-1H-pyrazole} palladium(II) (2)
L2 (139 mg, 0.771 mmol) and [PdCl2(NCMe)2] (100 mg, 0.386 mmol) were dissolved in CH2Cl2 (20 mL), and then stirred continuously at room temperature for 24 h, affording an orange solution. This was followed by in vacuo removal of the solvent to produce compound 2 as a yellowish-orange solid. Yield: 150 mg (72 %); melting point: 220-224 °C (decompose without melting). 1H NMR (400 MHz, CDCl3): Ô (ppm) 1.00 (s, 2x9H, 6xCH3); 1.76 (s, 2x9H, 6xCH3); 5.82 (s, IH, 4-H pz); 11.65 (s, 1H, N-H) (Fig. SI-10). 13C{1H} NMR (100 MHz, CDCl3) (ppm): 165.0 (Cf-C); 156.7 (Ce-C); 101.7 (Cd-C); 32.4 (Q-Ç); 31.1 (C-C); 30.0 Ca-CH3) (Fig. SI-11). Elemental analysis; Anal. calcd. for C22H40Cl2N4Pd: C, 49.12 %; H, 7.50 %; N, 10.42 %. Found C: 48.92 %; H, 7.16 %, N, 10.00 %.
2.2.3. Synthesis of Dichloro{bis-5-ferrocenyl-1H-pyrazole} palladium(II) (3)
L3 (90 mg, 0.3571 mmol) and [PdCl2(NCMe)2] (46 mg, 0.1785 mmol) were dissolved in CH2Cl2 (20 mL), followed by continuous stirring at room temperature for 24 h to produce an orange solution. Upon removal of the solvent in vacuo, compound 3 was obtained as a yellow-orange solid. Yield: 120 mg (49 %); melting point: 230-232 °C (decompose without melting).1H NMR (400 MHz, CDCl3): á(ppm) 7.89 (s, 1H, pz); 6.15 (s, 1H, pz); 4.57 (s, 2H, n5-C5H5); 4.34 (s, 2H, n5-C5H5); 4.14 (s, 5H, n5-C5H5); 11.53 (s, 1H, N-H) (Fig. SI-12). 13C{1H} NMR (100 MHz, CDCl3) (ppm): 144.8 (Cd-CH); 142.7 (Cc-CH); 103.7 (Cb-C); 71.6, 70.3, 67.0 (n5-CaH5) (Fig. SI-13). Elemental analysis; Anal. calcd. for C26H24Cl2Fe2N4Pd: C, 45.82 %; H, 3.55 %; N, 8.22 %. Found C, 45.76 %; H, 3.46 %; N, 8.27 %.
2.2.4. Synthesis of Dibromo{bis-5-ferrocenyl-1H-pyrazole} nickel(II) (6)
L3 (70 mg, 0.2778 mmol) and [NiBr2 (DME)] (45 mg, 0.1388 mmol) were dissolved in 20 mL CH2Cl2, and then stirred continuously at room temperature for 24 h producing an orange-brown solution. The solution was then concentrated and dried under vacuum for6hto yield complex 6. Yield: 70 mg (70 %); melting point: 210-212 °C; IR (vmax/cm-1): 3218 (N-H); 1623 (C=C); 1589 (C=N). Elemental analysis; Anal. calcd. for C26H24Br2Fe2N4Ni: C, 43.21 %; H, 3.35 %; N, 7.75 %. Found: C, 42.95 %; H, 3.27 %; N, 7.63 %.
2.3. Molecular Structure Determination
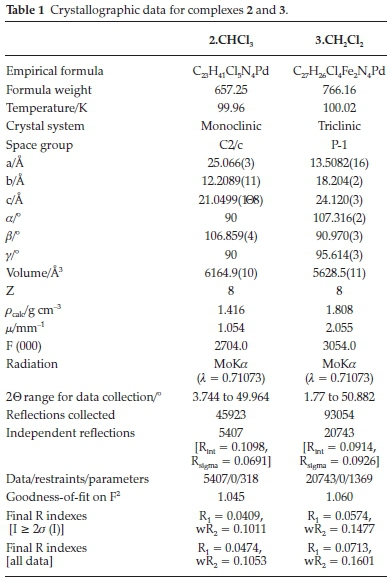
A mixture of CH2Cl2 (0.5 mL) and n-hexane (0.1 mL) for 2 or CHCl3 (0.5 mL) and n-hexane (0.1 mL) for 3 was used to obtain single crystals that were subsequently used for X-ray diffraction data collection for molecular structure determination.
Crystal data were collected using Bruker APEX-II CCD diffractometerwithMoKa (λ = 0.71073Ã).The diffractometerto crystal distance was 4.00 cm, and all crystal data collected at 100 K. Data reduction measurement was performed using SAINT+,21 and the intensity correction for absorption using SADABS.21 Refinement of structures, with least square minimization, was performed using the SHELXT22 and SHELXL23 software packages. All non-hydrogen atoms were refined with anisotropic displacement coefficients and placed in geometrically idealized positions, and constrained to ride on their parent atoms with relative isotropic coefficients.22,23
2.4. General Procedure for Hydrogenation Reactions
2.4.1. Hydrogenation with Molecular Hydrogen
Hydrogenation reactions using molecular hydrogen were studied in reactors with stainless steel vessels coupled with magnetic stirrers. In a typical experiment, the contents of the vessel consist of sorbic acid (0.5 mmol), catalyst (2.5 μmol, 0.5 mol%), hydrogen gas (5 bar) and methanol (5 mL). The solution mixture was purged twice with nitrogen gas, followed by the introduction of hydrogen gas (5 bar) and constant stirring of the mixture at 40 °C for 2 h. At the end of the reaction period, the reaction vessel was cooled, and the excess pressure generated was vented off slowly. The resulting hydrogenation products were withdrawn and filtered with MS nylon syringe filter (0.22 μm, 13 mm). Their % conversions were then determined by 1H NMR spectroscopy, using dioxane as an internal standard.
2.4.2. Hydrogenation with Formic Acid
In a typical experiment, sorbic acid (0.5 mmol), catalyst (2.5 μmol, 0.5 mol%) formic acid (20 mmol), KOH (4 mmol) and methanol (5 mL) were introduced into the reactor vessel. The solution mixture was purged twice with nitrogen gas, followed by stirring at 90 °C for 12 h. At the end of the reaction period, the reaction vessel was cooled, and the excess pressure generated was vented off slowly. The hydrogenation products were drawn out of the reactor vessel, filtered using MS® nylon syringe filter (0.22 μm, 13 mm) and their % conversions determined by 1H NMR spectroscopy, using dioxane as an internal standard.
3. Result and Discussion
3.1. Synthesis of Palladium and Nickel Complexes.
The pre-catalysts 1-6 were synthesized with compounds L1-L3 and the corresponding metal precursors, as shown in Scheme 1. Characterization of the palladium complexes was achieved using a combination of 1H NMR, IR, mass spectrometry and elemental analysis. The structures of the two new complexes, 2 and 3, were confirmed by single-crystal X-ray crystallography. Characterization of the nickel complexes, on the other hand, was carried out using mainly IR spectroscopy and elemental analysis. The structure of one of them, 4, was confirmed by single-crystal X-ray crystallography showing a structure result similar to that reported earlier.20
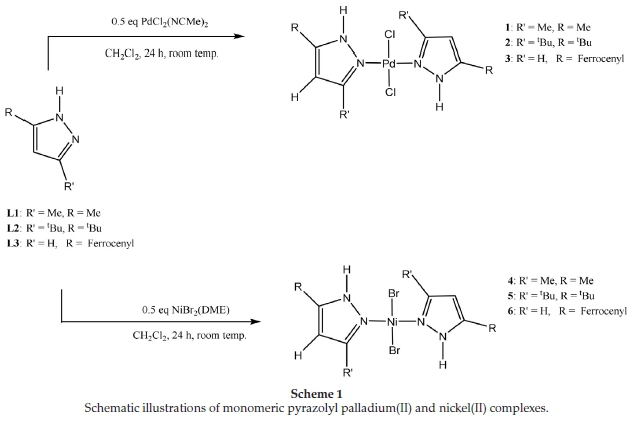
Characteristic chemical shifts of the pyrazolyl nitrogen protons (N-H) confirmed the successful ligand complexations. For instance, there was a downfield shift in the position of N-H proton from 11.20 ppm to 11.82 ppm in 1; from 10.19 ppm to 11.65 in 2; and from 10.62 ppm to 11.53 in 3. All other spectroscopic data were as expected and similar to those previously reported for 124,224, and 325.
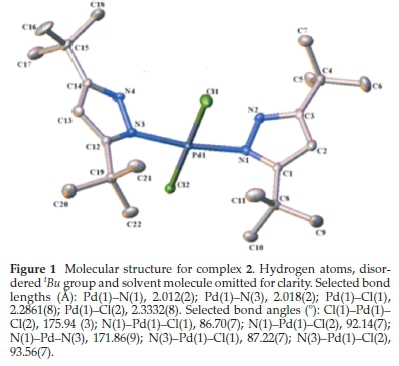
3.2. Molecular Structures of 2 and 3
Slow evaporation of solutions of 2 in CHCl3 and 3 in CH2Cl2 at room temperature produced orange crystals good enough for single-crystal X-ray crystallographic analysis. The crystals and structure refinement information are shown in Table 1, and the molecular structures in Figs. 1 and 2. Complexes 2 and 3 crystallized in C2/c and P-1 space groups, respectively. Figures 1 and 2 show that the pyrazole nitrogen atom is coordinated to the palladium in square planar geometries. However, the lBu groups in 2 are disordered. This disorder was handled during the refinement process by rotating the H atoms around the tBu axis to minimize the restraint caused by the tert-butyl groups. Selected bond distances and angles for the two palladium complexes are shown in the captions of Figs. 1 and 2. In 2.CHCl3the square planar geometry is distorted from 90 ° to the following bond angles: N(1)-Pd(1)-Cl(1), 86.70(7); N(1)-Pd(1)-Cl(2), 92.14(7); N(3)-Pd(1)-Cl(1), 87.22(7); N(3)-Pd(1)-Cl(2), 93.56(7)), whereas in 3.CH2Cl2there is just a slight distortion from the square planar geometry to the following bond angles: N(2)-Pd(1)-Cl(1), 89.41(12); N(2)-Pd-Cl(2), 89.10(12); N(4)-Pd(1)-Cl(1), 90.81(12); N(4)-Pd(1)-Cl(2), 90.70(12)). Similar distortions in square planar geometries of 2.CH2Cl2and 2.1/2Et2O have been observed by Li et al.24The average Pd-N bond lengths for 2.CHCl3and 3.CH2Cl2 are 2.105 (2) Â and 2.003 (4) Â, respectively. These average Pd-N bond lengths are longer in 2.CHCl3than the average Pd-N distance reported by Li et al.24for 2.CH2Cl2and 2.1/2Et2O (2.026(10) Â), but the reported average Pd-N distance is significantly shorter in 3.CH2Cl2. The average Pd-Cl bond lengths for 2.CHCl3and 3.CH2Cl2were found to be 2.3010 (8) Â and 2.303 (12) Â, respectively.
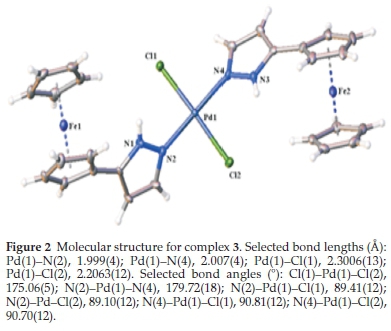
On the other hand, the nickel atom's geometry in 4 is a disordered tetrahedron where bond angles vary between 98.76(14) ° and 126.81(4) °. A similar distortion was reported by Nelana et al.20for the structure of [(3,5-Mepz)2NiBr2] where bond angles for the nickel complex vary between 98.90(11) ° and 125.94(4) °. Similarly, the average Ni-N bond lengths for 4 were 1.9695 (5) Â and 2.0669 (10) Â. The average Ni-Br bond lengths for 4 were also 2.3697 (10) Â and 2.4620 (5) Â. These data agrees with what was earlier reported.19 Selected bonds lengths and angles of 2 and 3 are stated under each molecular structure.
3.3. Catalytic Studies
3.3.1. Transfer Hydrogenation of Sorbic Acid
The pyrazolyl palladium and nickel complexes (1-6) were evaluated as catalysts for the transfer hydrogenation of sorbic acid, with formic acid as the source of hydrogen. Many phosphino nickel(II) and palladium(II) complexes are well-known catalysts for the hydrogenation of olefinic double bonds,26-29 some of them featuring PAN donor ligands.29,30 These hydrogenation reactions were first carried out in the presence of a base, such as KOH, which facilitates the deprotonation of the formic acid to formate ion. The formate ion then coordinates to the metal centre leading to decomposition of the formic acid with the aid of the intermediate, [M]-OOCH as an active catalyst, to produce H2 and CO2.29 This accelerates H2 heterolysis and causes the catalytic process.20(b),3°
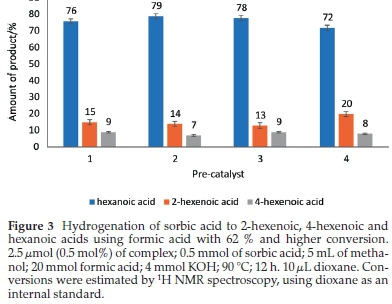
A typical hydrogenation reaction was performed with sorbic acid and a complex present in a 200:1 mole ratio at 90 °C for 12 h (Fig. SI-3). It is worth noting that the hydrogenation process did not take place when the pre-catalyst was not added. Catalytic activities (in terms of conversions of sorbic acid) are very good with the palladium complexes and in the order 2 > 3 >1; but poor for the nickel complexes, except for 4, which gave 62 % conversion (Table 2), having a turnover number (TON) of 124 and turnover frequency (TOF) of 10. However, the selectivity towards the distribution of the products is quite significant for all the complexes. Considering the effect of substituents (methyl, tertiary-butyl, and ferrocenyl) on complexes 1-3, the order of catalytic activity is 2 > 3 > 1 in terms of conversion of sorbic acids. Complex 2 gave 100 % conversion of sorbic acid in 12 h compared to 67 % and 74 % for complexes 1 and 3, respectively. A similar trend is observed in their TON and TOF values. However, there is not much difference in their selectivities towards 2-hexenoic and 4-hexenoic acids (Table 2, Fig. SI-4). The excellent activity of complex 2 might be due to the solubility of this compound provided by the tertiary-butyl substituent on the pyrazolyl ligand.
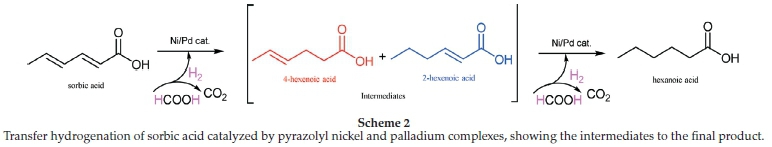
The pyrazolyl nickel and palladium catalyzed hydrogenation of sorbic acid, in this study, proceeds via a two-step (sequential) reaction. The first step involves the formation of the intermediates, 4-hexenoic and 2-hexenoic acids, and further hydrogenation of the hexenoic acid isomers in the second step produces hexanoic acid (Scheme 2).31
In an attempt to find out if the C=C bonds of the intermediates 4-hexenoic and 2-hexenoic acids would be fully saturated, the hydrogenation reaction was run for 24 h using complex 2. Interestingly, the product distribution after 24 h was only slightly different from when the reaction was run for 12 h, namely 82 % hexanoic acid, 13 % 2-hexenoic acid and 5 % 4-hexenoic acids for 24 h vs 79 % hexanoic acid, 14 % 2-hexenoic acid and 7 % 4-hexenoic acids for 12 h (Table 2, entries 2 and 3).
3.3.2.. Hydrogenation of Sorbic Acid with Molecular Hydrogen
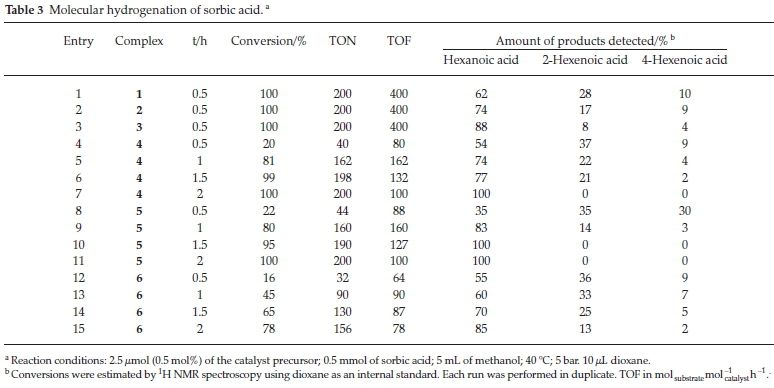
We also carried out the hydrogenation of sorbic acid with molecular hydrogen and complexes 1-6. In a typical experiment, 5 mmol of sorbic acid and 2.5 μmol (0.05 mol%) of the pre-catalysts were added to a reactor and ran for periods ranging from 0.5 h to 2 h, at 5 bar and 40 °C. The complex to sorbic acid ratio was 1:200, and the reaction was with palladium catalysts monitored by 1H NMR spectroscopy. All six complexes produced active catalysts, leading to the same products observed with formic acid (Scheme 2); the product distribution from the molecular hydrogenation reactions depicted in Table 3 is not so different from that of the formic acid reactions shown in Table 2 and Fig. 3.
As expected, the palladium complexes (1-3) are the most active, having the highest TOF of 400, all with complete conversion of sorbic acid within 0.5 h. Only two of the nickel complexes (4 and 5) had a complete conversion, and only after2h(Table 3, entries 7 and 11). Reactions, where the catalyst was a palladium complex, could be followed by 1H NMR spectroscopy (Fig. 4). Furthermore, all the nickel and palladium catalysts did not completely hydrogenate the sorbic acid to hexanoic acid within 0.5 h, and selectivities for 2-hexenoic and 4-hexenoic acids were as high as 37 % and 30 %, respectively (Table 3). These results indicate the conditions under which any of these intermediate products can optimally be obtained if partial hydrogenation compounds are the targeted products. This product distribution is depicted in Table 3 and in Fig. 4, which shows the 1H NMR spectral time study with complex 2.
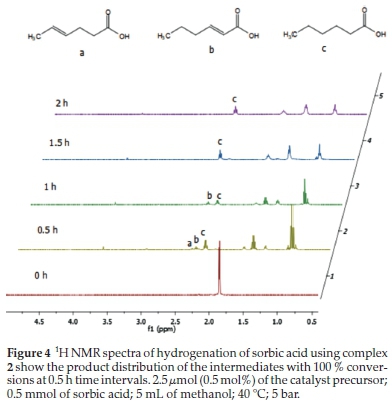
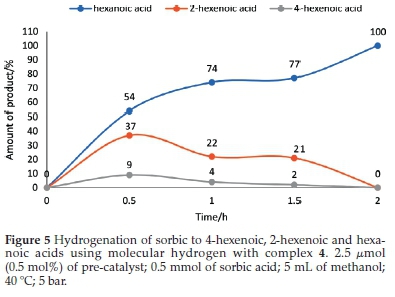
At 0.5 h, 20 % conversion of sorbic acid was observed with complex 4 (Fig. 4). This conversion resulted in 37% and9% selectivity towards 2-hexenoic acid and 4-hexenoic acid, respectively (Table 3, entry 4). After 1.5 h, the conversion was greatly increased to 99 % with 21 % selectivity towards 2-hexenoic acid, and2%of4-hexenoic was detected (Table 3, entry 6). Further increment in the reaction time (after 2 h) only formed hexanoic acid with 100 % conversion of sorbic acid (Table 3, entry 7). Similar observations were also seen using complexes 5 and 6 with 22 % and 18 % conversions, respectively, at 0.5 h.32 Results for the catalytic tests are summarized in Table 3. Our results clearly show that the less hindered C=C bond in 4-hexenoic acid is preferentially reduced over the more hindered one in 2-hexenoic acid. A detailed time-dependence study employing 1H NMR spectroscopic technique was carried out on a sample of complex 2 with 100 % conversion of sorbic acid (Fig. 4). The hydrogena-tion reaction's progress, with complex 2, produced a better-resolved spectrum in the chemical shifts compared to its nickel analogue (complex 5), which produced a broad spectrum due to its paramagnetic property. The hydrogenation reaction proceeded with the formation of the intermediates (2-hexenoic and 4-hexenoic acid) (Fig. SI-6) and hexanoic acid after 0.5 h. It is noted that by this time (0.5 h), the sorbic acid had been completely converted in all cases, and no traces of the substrate detected. Further increment in the time consumed one of the intermediates (4-hexenoic acid), followed by 2-hexenoic acid until all the C=C bonds were fully saturated after 2 h with 100 % selectivity towards hexanoic acid (Fig. 5).
4. Conclusions
Our study has shown how the nature of metal centres in pre-catalysts can affect hydrogenation reactions. This influence is demonstrated by the higher efficiency of (pyrazolyl)palla-dium(II) complexes as compared to their corresponding nickel(II) counterparts. The activities of the catalysts with the same ligand are: 1 > 4, 2 > 5 and 3 > 6. Furthermore, all the complexes investigated as sorbic acid hydrogenation catalysts, 1-6, gave appreciable conversions for catalytic hydrogenation of sorbic acid (i.e. greater than 52 %) compared with what was reported in the literature using ruthenium32,33 and rhodium.33 The product distributions for the hydrogenation of sorbic acid using pre-catalysts (1-4), with ones with significant conversions, is shown in Fig. 3.
Acknowledgements
The authors are grateful to the Royal Society-Department for International Development, United Kingdom (RS-DFID-UK) (Registered Charity Number 207043) for funding. We also thank Dr Banele Vatsha and Dr Gershon Amenuvor for assisting in solving the crystal structures, data collections and X-ray analysis.
Supplementary Material
Supplementary information is provided in the online supplement, and additional data can be obtained free of charge from The Cambridge Crystallographic Data Centre at the addresses below. CCDC numbers 1878431 and 1878429 contain the supplementary crystallographic data for complexes 2 and 3, respectively. Contact details are: Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (deposit@ccdc.cam.ac.uk or https://www.ccdc.cam.ac.uk/structures/
ORCID iD
O.E. Olaoye: orcid.org/0000-0002-7236-2823
References
1 (a) G. Hoge, Synthesis of both enantiomers of a p-chirogenic 1,2-bisphospholanoethane ligand via convergent routes and application to rhodium-catalysed asymmetric hydrogenation of CI-1008 (Pregabalin), J. Am. Chem. Soc., 2003, 125, 10219-10227; (b) K.B. Hassen, Y. Hsiaso, F. Xu, N. Ikemoto, Y. Sun, F. Spindler, C. Malan, E.J.J. Grabowski and J.D. Armstrong, Highly efficient asymmetric synthesis of sitagliptin, J. Am Chem. Soc., 2009,131,8798-8804; (c) H.U. Blaser, B. Pugin, F. Spindler andM. Thommen, From a chiral switch to a ligand portfolio for asymmetric catalysis, Acc. Chem. Res., 2007, 40, 1240-1250; (d) L.A. Saudan, Hydrogenation processes in the synthesis of perfumery ingredients, Acc. Chem. Res., 2007,40, 1309-1319. [ Links ]
2 F.H. Jardin, (S.J. Lippard ed.), Chlorotris(triphenylphosphine)rhodium(I): its chemical and catalytic reaction, Prog. Inorg. Chem., 1981, 28, 63-202. [ Links ]
3 J.A. Osborn, F.H. Jardine, J.F. Young and G. Wilkinson, The preparation and properties of tris(triphenylphosphine)halogenorhodium(I) and some reactions thereof including catalytic homogeneous hydrogenation of olefins and acetylenes and their derivatives, J. Chem. Soc., 1966 (A), 1711-1731. [ Links ]
4 R.A. Farra-tobaz, Z. Wei, H. Jiao, S. Hinze and J.G. De Vries, Selective base-free transfer hydrogenation of a,ß-unsaturated carbonyl compounds using z'PrOH or EtOH as hydrogen source, Chem. Eur. J., 2018, 24, 2725-2734. [ Links ]
5 (a) S. Bauri, S.N.R. Donthireddy, P.M. Illam and A. Rit, Effect of ancillary ligand in cyclometalated Ru(II)-NHC-catalyzed transfer hydrogenation of unsaturated compounds, Inorg. Chem., 2018, 57, 14582-14593; (b) S. Naskar and M. Bhattacharjee, Regiospecific solvent-free transfer hydrogenation of a,ß-unsaturated carbonyl compounds catalysed by a cationic ruthenium(II) compound, Tetrahedron Lett, 2007,48, 465^67. [ Links ]
6 (a) P. Wang, H. Liu, M. Liu, R. Li and J. Ma, Immobilized Pd complexes over HMMS as catalysts for Heck cross-coupling and selective hydro-genationreactions, New J. Chem., 2014,38,1138-1143; (b) G. Shang, W Li and Z. Zhang, in Catalytic Asymmetric Synthesis, (I. Ojima ed.), vol. 1,3rd edn., John Wiley and Sons, New York, USA, 2010, pp. 343-436. [ Links ]
7 Y. Liu, I.D. Gridnev and W. Zhang, Mechanism of the asymmetric hydrogenation of exocyclic a,ß-unsaturated carbonyl compounds with an iridium/BiphPhox catalyst: NMR and DFT studies, Angew. Chem. Int. Ed., 2014, 53, 1901-1905. [ Links ]
8 F. Nerozzi, Heterogeneous catalytic hydrogenation, Plat. Met. Rev., 2012, 56, 236-261. [ Links ]
9 (a) E. Bouwman, Handbook of Homogeneous Hydrogenation, (J.G. De Vries and C.J. Elsevier eds.), Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim, 2007, p. 93; (b) Z.R. Dong, Y.Y. Li, S.L. Yu, G.S. Sun and J.X. Gao, Asymmetric transfer hydrogenation of ketones catalysed by nickel complex with new PNO-type ligand, Chin. Chem. Lett., 2012, 23, 533-536; (c) Y.Y. Li, S.L. Yu, W.Y. Shen and J.X. Gao, Iron-, cobalt-, and nickel-catalysed asymmetric transfer hydrogena-tion of ketones, Acc. Chem. Res., 2015, 48, 2587-2598. [ Links ]
10 R.J. Liua, P.A. Croziera, C.M. Smith, D.A. Huculc, J. Blacksond and G. Salaita, Metal sintering mechanisms and regeneration of palladium/alumina hydrogenation catalysts, Appl. Catal A Gen., 2005, 282, 111-121. [ Links ]
11 (a) M. Shevlin, M.R. Friedfeld, H. Sheng, N.A. Pierson, J.M. Hoyl, L.C. Campeau and P.J. Chirik, Nickel-catalyzed asymmetric alkene hydrogenation of a,ß-unsaturated esters: high-throughput experimentation-enabled reaction discovery, optimisation, and mechanism elucidation, J. Am. Chem. Soc., 2016, 138, 3562-3569; (b) A. Corma, S. Iborra and A. Velty. Chemical routes for the transformation of biomass into chemicals, Chem. Rev., 2007, 107, 2411-2502. [ Links ]
12 (a) E. Negishi, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley and Sons, New York, 2002,1,229-247; (b) F.A. Harraz, S.E. El-Hout, H.M. Killa and I.A. Ibrahim, Palladium nanoparticle stabilised by polyethylene glycol: efficient, recyclable catalyst for the hydrogenation of styrene and nitrobenzene, J. Catal., 2012, 286, 184-192. [ Links ]
13 B. Ding, Z. Zhang, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Chemoselective transfer hydrogenation of a,ß-unsaturated ketones catalysed by pincer-Pd complexes using alcohol as a hydrogen source, Org. Lett., 2013,15, 3690-3693. [ Links ]
14 A. Bacchi, M. Carcelli, M. Costa, A. Leporati, E. Leporati, P. Pelagatti, C. Pelizzi and G. Pelizzi, Palladium(II) complexes containing a P,N chelating ligand part II. Synthesis and characterisation of complexes with different hydrazinic ligands. Catalytic activity in the hydrogenation of double and triple C-C bonds, J. Organomet. Chem., 1997,535, 107-120. [ Links ]
15 P.W.N.M. Leeuwen and C.J. Chadwick, Homogeneous Catalysts: Activity-Stability-Deactivation, Wiley-VCH Verlag, Germany, 2011. [ Links ]
16 M.W. Van Laren and C.J Elsevier, Selective homogeneous palladium(0)-catalysed hydrogenation of alkynes to (Z)-alkene. Angew. Chew. Int. Ed., 1999, 38, 3715-3717. [ Links ]
17 J. Elguero and E.J.R. Jacquier, Azoles VII, Assignment of the nuclear magnetic resonance signals of N-substituted pyrazole, Bull, Soc. Chim. Fran. 1968, 2, 707-711. [ Links ]
18 K. Niedenzu, J. Sorwatowski and S. Trofimenko, Boron derivatives of 3-ferrocenylpyrazole, Inorg. Chem., 1991, 30, 524-527. [ Links ]
19 (a) G.K. Anderson andM. Lin, Bis(benzonitrile)dichloro complexes of palladium and platinum, Inorg. Synth., 1990,28,60-63: (b) E. Ocansey, J. Darkwa and B.C.E. Makhubela, Bis(pyrazolyl)palladium(II) complexes as catalyst for Mizoroki-Heck cross-couplingreactions, Polyhedron, 2019,166, 52-59. [ Links ]
20 (a) S.M. Nelana, J. Darkwa, I.A. Guzei and S.F. Mapolie, Ethylene polymerisation catalysed by substituted pyrazole nickel complexes, J. Organomet. Chem., 2004, 689, 1835-1842; (b) G. Amenuvor, B.C.E. Makhubela and J. Darkwa, Efficient solvent-free hydrogenation of levulinic acid to y-valerolacetone by pyrazolylphosphite and pyrazolylphosphinite ruthenium(II) complexes, ACS Sustainable Chem. Eng., 2016, 4, 6010-6018. [ Links ]
21 APEX2, (including SAINT SADABS), Bruker AXS Inc., Madison, WI, 2012. [ Links ]
22 G.M Sheldrick, SHELXT- Integrated space-group and crystal structure determination, Acta Crystallogr. Sect. Found. Adv., 2015, A71,3-8. [ Links ]
23 G.M. Sheldrick, Crystal structure refinement with SHELXL, Acta Cryst., 2015, C71, 3-8. [ Links ]
24 K. Li, J. Darkwa, I.A. Guzei and S.F. Mapolie, Synthesis and evaluation of substituted pyrazoles palladium(II) complexes as ethylene polymerisation catalysts, J. Organomet. Chem., 2002, 660, 108-115. [ Links ]
25 C. Obuah, A. Munyaneza, I.A. Guzei and J. Darkwa, Ferrocenyl-pyrazolyl palladium complexes as catalysts for the polymerisation of 1-heptene and 1-octene to highly branched polyolefins, Dalton Trans., 2014,43, 8940-8950. [ Links ]
26 (a) I.M. Augulo, E. Bouwman, R. Gorkum, S.M. Lok, M. Lutz and A.L. Spek, New nickel-containing homogenous hydrogenation catalysts structures of [Ni(o-MeO-dppol)Cl2] and [Ni(dcpe)Cl2], J. Mol. Cal., 2003, 202, 97-106; (b) Y. Liu, Z. Yi, X. Tan, X-Q. Dong and X. Zhang, Nickel-catalyzed asymmetric hydrogenation of cyclic sulfamide imines: efficient synthesis of chiral cyclic sulfamidates, iScience, 2019, 19, 63-73. [ Links ]
27 A. Togni, C. Breutel, A. Schnyder, F. Spindler, H. Landert and A. Tijiani, A novel easily accessible chiral ferrocenyldiphosphine for highly enantioselective hydrogenation, allylic alkylation, and hydrogenation reactions, J. Am. Chem. Soc., 1994,116, 4062-4066. [ Links ]
28 Y. Hamada, Y. Koseki, T. Fujii, T. Maeda, T. Hibino and K. Makino, Catalytic asymmetric hydrogenation of a-amino-ß-keto ester hydro-chlorides using homogeneous chiral nickel nickel-bisphosphine complexes through DKR, Chem. Comm., 2008, 46, 6206-6208. [ Links ]
29 C. Pérez-Zúñigaa, C. Negrete-Vergarac, V Guerchaisb, H. Le Bozecb, S.A. Moyaa and P. Aguirre, Hydrogenation of N-benzylideneaniline by palladium(II) catalysts with phosphorus-nitrogen ligands using formic acid as a renewable hydrogen source, Mol. Catal., 2019, 462, 126-131. [ Links ]
30 S.M. Lu, Z. Wang, J. Li, J. Xiao and C. Li, Base-free hydrogenation of CO2 to formic acid in water with an iridium complex bearing a N, N'-diimine ligand, Green Chem., 2016,18, 4553-4558. [ Links ]
31 J. Heinen, M.S. Tupayachi, B. Drießen-Hölscher, Biphasic homogeneous hydrogenation of sorbic acid with water-soluble ruthenium catalysts: aspects of mass transfer, Catalysis Today, 1999, 48, 273-278. [ Links ]
32 B. Drießen-Hölscher and J. Heinen, Selective two-phase hydrogenation of sorbic acid with novel water-soluble ruthenium complexes, J. Organomet. Chem., 1998, 570, 141-146. [ Links ]
33 A. Udvardy and Á. Kathó, Hydrogenation of sorbic acid in mono and biphasic systems catalysed by Rh(I)-phosphine complexes, React. Kinet. Catal. Lett., 2008, 95, 81-87. [ Links ]
Received 6 March 2020
Revised 31 December 2020
Accepted 20 January 2021
* To whom correspondence should be addressed. E-mail: O.O., oyetunjl@ub.ac.bw / J.D., jdarkwa@bltrl.co.bw
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]


{kind=link}
{kind=link}
{kind=link}