










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.75 Durban 2021
http://dx.doi.org/10.17159/0379-4350/2021/v75a5
RESEARCH ARTICLE
https://doi.org/10.17159/0379-4350/2021/v75a5
Ethylene Trimerization over Supported SNS and PNP
Chromium Catalysts; Mzamo L. Shozi; Xolelwa Zulu; Holger B. Friedrich*
School of Chemistry and Physics, University of KwaZulu-Natal, Durban, 4000, South Africa
ABSTRACT
Chromium(III) complexes with sulphur, nitrogen and phosphorus tridentate ligands were synthesized and characterized. These complexes were supported on SiO2 and characterized by BET surface area measurements, XRF, SEM-EDX and FTIR. The complexes were tested for activity and selectivity in the trimerization of ethylene. The substituent's effect and influence on the sulphur on the supported catalysts were studied using the ethyl and the decyl substituted catalysts. The influence of temperature on catalytic performance was evaluated using the PPP supported system. The most active supported catalyst, the decyl substituted SNS catalyst, showed good activity of up to 19 500 g/g Cr h-1 and selectivity of 97.3 % to C6 products (98.2 % 1-hexene). This activity and selectivity were comparable to the homogeneous counterpart's performance that achieved 22 000 g/g Cr h-1 and 98.2 % C6 products (96.7 % 1-hexene), which surpassed the ethyl substituted catalyst, which was not supported, under the same reaction conditions. The supported PPP catalyst activities showed it was sensitive to higher temperatures, but this depends on the supporting technique.
Keywords: Ethylene trimerization, sulfanyl ligands, phosphine ligands, supported catalysts.
1. Introduction
Supported metal complexes that are well known for ethylene conversions are metallocenes, which are immobilized for ethylene polymerization reactions.1,2 As a result of the different routes of immobilization and strategies that have been proposed, different catalyst activities and/or polymer properties are obtained. The support alters the supported catalyst activity due to steric and electronic effects attributable to the support itself.3-8 In many catalytic systems, the trend that has been observed is that the supported complexes exhibit lower catalytic activities than their homogeneous counterparts. This decrease in the activities can be attributed to many factors, viz. (i) deactivation of the metal complexes during the grafting reaction, (ii) inaccessibility of the metal complex to the co-catalyst thus hindering the process of catalyst activation, (iii) generation of active sites that have slow propagation rates due to interactions with the support surface and (iv) restrictions of the monomer access to the active site preventing the chain growth.9 Another complexity that encumbers these kinds of systems is the high levels of heterogeneity of the metal sites formed on, for example, the amorphous silica surface together with the high dilution of the metal. The well-known Cr(II)/SiO2 Phillips catalyst, though the most successful commercially, is among the least understood at a molecular level.10
These challenges have not stopped scientists from studying these kinds of systems. Two of the common ways of recycling the 'homogeneous' catalyst from the product phase are two-phase11,12 and immobilization catalysis13-17. Although studied extensively, the latter has had little commercial success except for the Phillips catalyst.18-20 The bonding of the silica to the metal complexes can be described in three ways: the metal complexes can be directly adsorbed on the surface of the support2,21, the metal complexes can be synthesized in situ on the support, or the silica can be chemically modified with methylaluminoxane, alkyl aluminium borates and organosilanes.4
Although some researchers have noted that homogeneous catalysts are much more active and selective than their heterogeneous counterparts22-24, Monoi and Sasaki25 reported a new, highly active and selective silica-supported ethylene trimeriza-tion catalyst. This catalyst system was obtained by supporting tris[bis(trimethylsilyl)amido]-Cr(III)[N(SiMe3)2]3 and isobutyl-aluminoxane on silica, which has been calcined at 600 °C. The catalyst loading was 1.0 wt % Cr with the activator/Cr molar ratio at 24, which gave 74.2 % 1-hexene selectivity. Nenu and Weckhuysen26 discovered another supported system that proved successful for trimerization of ethylene. This catalyst that consisted of a Cr centre attached via two silanol groups to an amorphous silica carrier was more active than its homogeneous parallel. The 1,3,5-tribenzylhexahydro-1,3,5-triazine (TAC) ligand was anchored on a Cr/SiO2 surface. This ligand was reported to form active trimerization complexes with different metals. At a Cr/TAC ratio of 1, the catalyst was observed to present predominantly trimerization centres. The catalyst also showed single-site behaviour, which was confirmed by the low value of the PI (polydispersity index) obtained from gel permeation chromatography measurements of the polymer produced. The TAC ligand's coordination was reported to narrow down the diversity of the Cr active sites previously on the silica surface. The required reaction conditions for ethylene conversion by this heterogeneous system were low temperatures and pressures. At elevated temperatures, the catalyst exhibited polymerization activity with less 1-hexene formed.26
This study reports the use of supported sulfanyl and phosphine chromium complexes for the trimerization of ethylene, which has not been previously reported. Homogeneous sulfanyl and phosphine catalysts have been reported to give good results, so we hoped to improve these by supporting them or gaining a separation advantage.
2. Experimental
2.1. Materials and Instrumentation
Unless otherwise stated, all reactions were carried out using conventional Schlenk techniques under an inert atmosphere of pre-dried nitrogen. THF was dried and distilled under an inert atmosphere using sodium and benzophenone. Diethyl ether was dried over sodium wire before use. Melting points were obtained using an Ernst Leitz Wetzlar Hotstage Microscope. Elemental analyses of the complexes were conducted using a LECO CHNS elemental analyser. NMR spectra were recorded on a Bruker Avance III 400 MHz spectrometer and a Bruker Avance III 600 MHz solid-state MAS spectrometer fitted with a 4 mm MAS BB/1H probe.
These chemicals were commercially available and they were used as received: butanethiol (purum > 97 % GC) (Fluka), 1-decanethiol (techn purum > 95 % GC) (Fluka), ethanethiol (purum > 97 % GC) (Fluka),bis(2-chloroethyl)amine hydrochloride 98 % (Aldrich), triethylamine (puriss > 99.5 % ) (Fluka), DMSO (puriss, standard for GC), AIBN 98 % (Acros Organics), SiO2 fumed, 99.8 % (Aldrich), lithium sheets, triphenyl-phosphine 99 %, (Aldrich), ammonium chloride (>99.5 %) (Aldrich), trimethylsilyl chloride, divinylphenylphosphine, Cr(Cl)3(THF)3 97 % (Aldrich), NaOH (pellets, Rochelle Chemicals), HNO3 (65 %, AAR, SMM Instruments), diethyl ether (Merck, 99 % ), HCl (fuming, 37 % for analysis) (Merck), potassium tert-butoxide (purum, 1 M in THF) (Fluka).
Ethylene was supplied by Air Liquide and passed through an Al2O3 guard-bed. MMAO-3A (Akzo Nobel) was used as received. Ethanol (absolute, 99.5 % ) was used as received from Merck. Toluene (for analysis) (Merck) and methylcyclohexane (for synthesis) (Merck) were percolated under nitrogen through an Al2O3 column. The GC standards, 1-hexene (Aldrich, 97 % ), 1-octene (Sigma-Aldrich, 98 % ), nonane (Sigma-Aldrich, 99 % ) and methylcyclopentane (Fluka, 95 % ) were used as received. GC-FID analyses were carried out on a Perkin Elmer Clarus 500 chromatograph using a PONA 50 m x 0.32 mm x 0.50 μm column, oven programme: Initial temperature = 40 °C, hold for 20 min, ramp at 4 °C/ min-1 to 300 °C and hold for 5 min, carrier gas: nitrogen.
2.2. Catalyst Synthesis
2.2.1. Homogeneous Catalysts
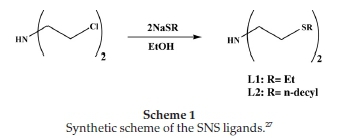
2.2.1.1. Synthesis of Bis[2-(ethylsulfanyl)]amine Ligands (L1-L2) Bis[2-(ethylsulfanyl)]amine ligands L1-L2 were synthesized according to literature procedures or modification thereof (Scheme 1).27 Full details are given in the Supplementary Material.
2.2.1.2. Synthesis of Bis[(2-diphenylphosphino)ethyl]amineHCl (PNP)
Bis[(2-diphenylphosphino)ethyl]amine-HCl ligand was synthesized according to a modified literature procedure (Scheme 2).28 Full details are given in the Supplementary Material.

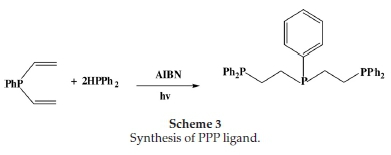
2.2.1.3. Synthesis of Bis(2-diphenylphosphinoethyl)phenylphosphine Ligand (L3)
In a sealed Schlenk tube, a mixture of divinylphenylphosphine (0.165 g, 1.02 mmol), diphenylphosphine (0.38 g, 2.04 mmol) and AIBN (100 mg) was irradiated using a 350W Hg Arc lamp at room
temperature for 48 h (Scheme 3). The resulting white solid was washed with n-pentane, and the excess volatiles were removed under reduced pressure. Yield (0.49 g, 90.7 % ). 1H NMR aH(CDCl3): 1.4-1.6 (8H, m), 7.2-7.6 (25 H,m) 13C NMR δC(CDCl3): 25.2 (CH2CH2-PPh2) aromatic C (128-132). 31P NMR (CDCl3): 20 d (P-P). Mp 129-130 °C. Elemental analysis (EA): C34H33P3; C, 76.39; H, 6.22. Found C, 76.36; H, 6.34.
2.2.1.4. Synthesis of Chromium Complexes of Bis[2-(ethylsulfanyl)] amine (1) and Bis[2-(decylsulfanyl)]amine Ligands (2)
Complexes 1 and 2 were synthesized according to literature procedures or modification thereof. Full details are given in the Supplementary Material.27
2.2.1.5. Synthesis of Chromium Complex of Bis[(2-diphenylphos-phino)ethyl]amine Ligand (Cr-PNP)
Cr-PNP was synthesized according to literature procedures or modification thereof. Full details are given in the Supplementary Material.28
2.2.1.6. Synthesis of Chromium Complex of Bis(2-diphenylphos-phinoethyl)phenyl-phosphine Ligand (3)
CrCl3(THF)3 (0.372 g, 1 mmol) was dissolved in dichloro-methane and filtered. Bis(2-diphenylphosphinoethyl)phenyl-phosphine (0.535 g, 1 mmol) was dissolved in dichloromethane (40 mL) and added to the filtrate under nitrogen. The solution was then stirred at room temperature. The solvent was removed under reduced pressure, and the solid product was left stirring overnight in petroleum ether. The blue solid was isolated by filtration. Yield (0.275 g, 45.8 % ). IR (KBr) cm-1 = P-C 1133 (s); Cr-P 688 (s); Cr-Cl478 (m). EA= C34H33Cl3CrP3: C, 58.94; H, 4.80.
Found C, 58.76; H, 4.75.
2.2.2. Heterogeneous Catalysts
2.2.2.1. Incipient Wetness Impregnation Method (Supported Complexes-SC) (SC1-SC3)
Silica (SiO2) was pre-calcined in a stream of dry N2 at 600 °C for 24 h. A pre-determined amount of the support (SiO2) was placed in a round-bottomed flask and solvent (toluene) was added.
After warming to ~100 °C, to give a Cr loading of -1 %, an appropriate amount of the chromium complex in solution was added to the support. The two were mixed at -115 °C. After stirring for 2 h, the toluene was removed in vacuo at 120-130 °C under reduced pressure. The catalysts were obtained as bright green or bright blue powder depending on the supported complexes. The sulphur-based complexes gave green powders, and phosphorus-based complexes gave blue powders. The catalysts were left to dry at 40 °C overnight.
2.2.2.2. Two-step Incipient Wetness Impregnation Method (Supported Ligands-SL) (SL1-SL3)
The silica was first impregnated with the ligand's solution (in toluene) of a known concentration. Thus, the silica and the ligand were mixed and stirred at -100 °C for 2 h. This mixture was then dried (solvent evaporated) at 115 °C. The dried solid product was impregnated for a second time with a solution of a chromium precursor, CrCl3(THF)3, to obtain a 1.0 wt % chromium loading. This mixture was stirred again for2hat -115 °C. The solvent was then removed under reduced pressure at 120-130 °C. The catalysts were obtained as light green and bright green powders. The catalysts powders were dried at 40 °C overnight.
2.3. Catalyst Testing
AParr® reactor, fitted with a gas entrainment stirrer and internal cooling coil, was used with water as a coolant. It was manually controlled to maintain the desired temperature. The reactor allowed for the charging of the catalyst solutions and the co-catalyst (MMAO-3A) under pressure, which in turn allowed for catalyst activation in the presence of ethylene. A mass flowmeter was used to monitor the ethylene uptake and generate ethylene uptake profiles with reaction time. The reaction pressure was chosen by setting the ethylene cylinder supply regulator, allowing the ethylene flow to vary on-demand, and maintaining constant pressure as it was consumed in the reactor through a flowmeter control. All heterogeneous catalytic tests were done in a slurry system using toluene as a suspension medium. The reaction time was 0.5 h for all experiments. The CrCl3-TAC homogeneous system was tested under identical reaction conditions using a different co-catalyst as in literature,26 i.e. MMAO-3A was used instead of the combination of N,N-dimethylanilinium tetra(pentafluoro-phenyl) borate (DMAB) and tri-isobutyl aluminium reported. A continuous flow of the ethylene feed was maintained throughout the reaction to maintain the reaction vessel's pressure. Care was taken to exclude the presence of air, and possible deactivation of catalysts was also reduced by using deoxygenated solvents. Catalytic leaching tests were performed using each catalyst: the supported catalyst, toluene and MMAO-3A were added to a flask under N2. The mixture was allowed to stir for 20 min, after which it was filtered. The filtrate was added to the reactor with toluene with MMAO-3A. The reactor was then charged with ethylene. The reaction product was analysed by GC-FID. Also, ICP-OES, where possible, was run on the samples.
3. Results and Discussion
3.1. Synthesis and Characterization of the Catalysts
3.1.1. Homogeneous Catalysts
The synthesis of the SNS ligands, as shown in Scheme 1, is a simple equimolar reaction between sodium hydroxide and the respective thiols. The base (NaOH) abstracts the acidic protonof the primary thiol forming the sodium alkylthiolate. The alkylthiolates react with the bis(2-chloroethyl) amine hydro-chloride to give the desired ligands with the alkyl pendant chains attached from the sulphur donors coordinating to the central nitrogen donor atom. The successful synthesis ofligands L1-L3 was confirmed with the amine (N-H) resonance on the central nitrogen donor atom. The characteristic broad singlet was observed between 1.89-2.14 ppm. The resonances of the ethyl spacers between the central donor atom, the nitrogen and the sulphur donors were shifted downfield due to the nitrogen. The resonances were observed as far downfield as 2.92 ppm. This effect was also observed with the ethyl protons attached directly to the sulphur donors.
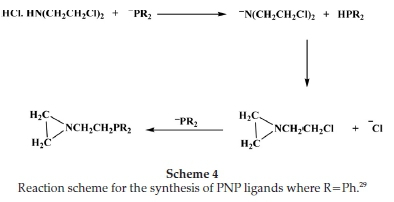
The structurally similar PNP analogues were obtained using two methods as described in the Supplementary Material. Method 1 employs the reductive cleavage of C-P bonds in triphenylphosphine by Na/NH3. The cleavage of triphenyl-phosphide by Na/NH3 gives sodium diphenylphoshine and sodium amide. Ammonium chloride is added to destroy the amide and concurrently convert the secondary phosphide to the diphenylphosphine. Although easy to manipulate, this method has low yields because the process is encouraged by the presence of electron-donating groups and since the target of this reaction is a pendant arm that is void of this, yields were low. In Scheme S1 (Supporting Information), the trimethylsilyl group's protecting function was used, since protic reagents readily cleave the nitrogen-silicon bonds. In the presence of excess trimethylchlorosilane, the amine hydrochloride reacts with triethylamine in DMSO.
In Scheme 4, the reaction of the amine chloride, bis(2-chloro-ethyl)amine hydrochloride, with the in situ prepared diphos-phide nucleophile is shown. The method is preferred over the liberation of the free amine first, followed by silylation, as this latter route results in low yields of mixtures of unidentified silylamines. The use of other silylation agents gave unsatisfactory results. An added advantage of this method of silylation was that, because the reaction was performed at -40 °C, lithium halogen exchange is minimized, and the hydrolysis of excess nucleophile and cleavage of the protecting function then occur concurrently.29
In an attempt to get higher yields and greater purity, the PNP ligand was isolated as a hydrochloride salt, as outlined in Scheme 2. The salt was obtained in a crystalline and air-stable form. The PPP ligand was synthesized as depicted in Scheme 3 due to a free-radical addition of P-H bonds to a carbon-carbon double bond using a radical initiator AIBN (2,2-azobisisobuty-ronitrile) in the presence of ultra-violet irradiation. The ligands were characterized by 1H, 13C and 31P NMR. The phosphorus-containing ligands PNP and PPP mostly displayed multiplets in the aromatic region, at 7.20-7.40 ppm. These resonances were attributable to the phenyl substituents on the phosphorus atoms. In the phosphorus-alkyl region, the methylene protons of the backbone both gave rise to multiplets. The shifts of these multiplets revealed that the protons on the carbons directly bonded to the nitrogen atom were more deshielded (2.753.20 ppm) relative to the protons on the carbons bonded to the phosphorus atom (1.38-1.61 ppm). Resonances for the N-H protons, in the phosphorus ligands, were not observed. Except in the case of PNP where the ligand was isolated as a salt. In this case, the amine protons were observed as a distinct singlet at 9.90 ppm.
3.1.2. Heterogeneous Catalysts
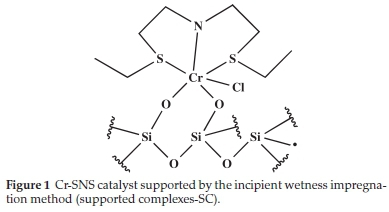
The SNS (ethyl and decyl) substituted and Cr-PPP complexes were supported on SiO2 by two methods. In the first method, the ligand is physisorbed onto the support, and the chromium precursor is then added; these are termed supported ligands (SL). The second method involved supporting the synthesized complexes on the support termed supported complexes (SC). The chromium in the complexes is paramagnetic, and therefore solution NMR could not be performed. The supported complexes were characterized by XRF, BET, SEM/EDX and FT-IR. Typical IR absorption bands of the metal-ligand bonds (the metal hetero-atom vibrations for the SL and SC material) were observed. The proposed interaction of the pre-formed complexes and the support is shown in Fig. 1, exemplified by the ethyl substituted SNS complex. The support had been calcined at high temperature to remove excess surface hydroxyl groups.
3.1.2.1. X-ray Fluorescence
The content and loading of the chromium on the surface of the support were determined using X-ray fluorescence (XRF). This method was chosen because of the limited solubility of SiO2 in organic acids. Also, because of the low metal content in the prepared catalysts, this method proved best. It has been reported that at metal loadings of 0.5-3.0-wt % the supported species are sufficiently separated from each other to give structures that act almost independently of each other.30 The targeted metal loading on the support surface was 1 % Cr/SiO2 (wt/wt). The actual metal loadings obtained for the differently synthesized catalysts are shown in Table 1.

3.1.2.2. Brunauer-Emmett-Teller (BET) Surface Area Measurements The BET surface area measurements are shown in Table 2. The results showed that all catalysts have lower surface areas than the neat support, calcined and uncalcined. The Cr(III)SNS with the ethyl substituent has the highest surface area, with its ligand supported version showing higher surface area measurements than the catalysts supported through the complex. The opposite is true for Cr(III)SNS with the decyl substituent, with the catalyst supported through the complex, showing the higher surface area measurements. This observation could be explained as a phenomenon of size and bulk of the substituents, rather than the nature of the interaction with the support. For example, the larger the molecules on the catalysts support surface, the more blocked the surface of the support is, the less open pores on the surface will be and thus the observed decrease in the surface area.
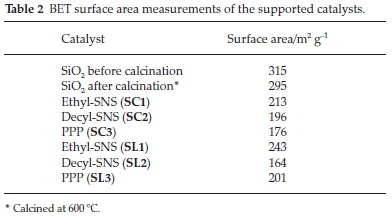
Noteworthy is that for the complexes 1 and 3, the route of supporting the ligand (SC1-SC3) yielded higher surface areas than obtained for the complex supported counterparts (SL1-SL3). The trend is reversed with the complex 2 substituted. An explanation for this could be that the less bulky substituents, e.g. ethyl, block pores less on the surface of the support and thus increased surface area measurements are obtained. When the complex is supported, it blocks more surface pores and decreases the surface area. With complex 2, the ligand is bulkier, and it blocks more surface pores, but with the complex, there are more surface pores exposed, and thus the surface area was higher with the catalysts supported through the complex.
3.1.2.3. Scanning Electron Microscopy-Energy Dispersive
Spectroscopy
Scanning electron microscopy-energy dispersive spectros-copy (SEM-EDS) was used to investigate the distribution and homogeneity of the elements on the surface of the support. Fig. 2 shows the SEM-EDS images of the supported catalysts. The catalysts prepared by the one-step incipient wetness impregnation method (SC1 and SC2), show that the metal is evenly distributed across the surface of the support. The sample prepared by the two step-incipient impregnation method (SL1 and SL2) showed that the complex has an uneven distribution on the surface of the support.

3.1.2.4. Infrared Spectroscopy
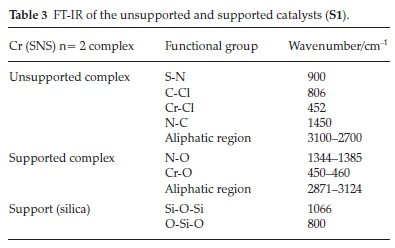
Silica traditionally has siloxane bridges (Si-O-Si), hydrogen bonded hydroxyl groups and free hydroxyl groups which can be either geminal or single (isolated).31 The IR data in Table 3 show the absence of the acidic silanol groups and the presence of siloxane groups. The absorption bands at 1066 and 800 cm-1 are assigned to the Si-O-Si bridges and the O-Si-O linkages, respectively. The characteristic peaks in the IR spectra for the supported catalysts are those of the interactions of the support and the dispersed metal species on the SiO2. The most important of these interactions is a result of the silica-metal interaction. In this case, the metal interacts with the oxygen atom of the silica bridges. The absorption band for the metal-oxo group (Cr-O) is observed at 450-160 cm-1 in all the catalysts. This absorption band is evidence for the replacement of one or two of the metal-halide bonds in the complexes. The catalysts that were prepared by the two-step incipient impregnation method showed similar absorption bands. With these catalysts absorption bands observed between 1344 and 1385 cm-1 and 1515 and 1560 cm-1 for the ethyl and decyl substituted catalysts, respectively, might imply an N-O interaction with the support. This suggests that the bonding of atoms in these catalysts is indeed different from those to the catalysts synthesized by the one step incipient impregnation method.
3.2. Catalyst Testing
3.2.1. Ethylene Trimerization using a Commercial Ligand
Nenu and Weckhuysen26 had success in ethylene trimerization with a tridentate supported Cr-TAC-complex. The ligand was used as a benchmark for application in our reaction system. The ligand 1,3,5-tribenzylhexahydro-1,3,5-triazine (TAC) was reacted with Cr(Cl)3(THF)3 to obtain a pre-catalyst Cr-TAC. Cr-TAC was subsequently supported on SiO2 to give a catalyst loading of ~1 wt % Cr/SiO2. Table 4 shows the catalytic performance of the homogenous and supported catalysts at both 30 and 40 bar ethylene pressure. Cr-TAC behaved as a polymerization catalyst as evident by the formation of almost exclusively polyethylene (PE) at both pressures. Upon increasing the pressure, the activity of the catalysts decreased and slightly more polymer formed, with decreasing selectivity to the C6 products. The amount of PE formed at both pressures is >79 %. 1-C6 (1-hexene) selectivity within the C6 fraction showed that the ethylene trimerization product was a minor co-product when the supported Cr-TAC complex was used. The catalytic activities were fairly low, with Cr-TAC achieving 3191 g/g Cr h-1 at 30 bar and the activity decreased when the pressure was increased to 40 bar, to 2793 g/g Crh-1.
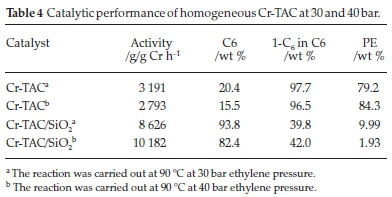
When the Cr-TAC catalyst was supported on SiO2, an interesting result was obtained. The supported catalyst showed more ethylene trimerization character (C6 formation) than its homogeneous counterpart. A very significant decrease in the amount of PE formed was noted. This decrease noted enhanced selectivity to 1-C6 could be due to the distortion of the Cr coordination induced by the support oxide.26 This also leads to higher selectivities at slightly increased pressures. Simultaneously an increase in the amount of the C6 fraction formed was found. At 30 bar, Cr-TAC/SiO2 only formed 10 % PE compared to 79 % PE formed by Cr-TAC, and at 40 bar Cr-TAC/SiO2 formed 2 % PE compared to 84 % formed by Cr-TAC. Noteworthy is the relatively low amount of 1-C6 within the C6 fractions (only 42 and 40 %, respectively). Other C6 products were major by products in this reaction under these conditions. These results were also encouraging in that the catalyst activity of Cr-TAC/ SiO2 increased, achieving 8626 g/g Cr h-1 at 30 bar and further increased at 40 bar to give 10 182 g/g Cr h-1.
3.2.2. Ethylene Trimerization using Synthesized Catalysts
3.2.2.1. Cr-SNS Catalysts
Table 5 shows the results of the homogeneous SNS catalysts with the ethyl (1) and decyl (2) substituents on the sulphur, together with their heterogeneous counterparts (SC1-SC2 and SL1-SL2). Catalyst 1 showed its highest activity of 8044 g/ gCrh-1 at 30 bar, compared to 40 bar where 7027 g/g Cr h-1 was achieved. Catalyst SC1 showed improved activity at both pressures. This improved catalyst activity was accompanied by the formation of less 1-C6 in the C6 fraction. Notably, the amount of PE formed by SC1 was less than that formed by 1. Catalyst SL1, however, showed lower activity than those of 1 and SC1. The C6 fraction was lower as well, comparing 98.6 % at 30 bar and 96.8 % at 40 bar achieved by SC1 to 90.9 % at 30 bar and 92.5 % at 40 bar achieved by SL1.
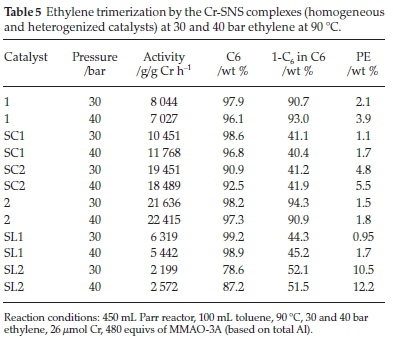
The influence of the support seems to favour catalyst activity, especially with the catalyst supported through the complex, SC1. The chromium centre is in direct contact with the support. The oxygen atoms decrease the electronic cloud around the metal and therefore, the nucleophilicity of the metal centre.26,32
This decreased electron cloud favours metallacycle growth in that it promotes substrate attack, in this case, ethylene insertion, and in turn, an increased C6 fraction. In terms of activity, the support seems to inhibit chain growth to heavier fractions, ultimately the undesirable PE. This activity is apparent with catalyst SC2, which behaves like the homogenous catalyst, and the support has the role of an inhibitor as activity decreases, and less C6 is formed. An increase in pressure favoured PE formation. The decrease in 1-C6 over SC1 is indicative that the disproportionation is favoured over reductive elimination of 1-C6.
Catalyst 2 showed increased activity that reached 21 636 g/g Cr h-1 at 30 bar and 22 414 g/g Cr h-1 at 40 bar. Upon supporting 2 on SiO2 to obtain SC2, the catalyst activity was lower at 19 451 g/g Cr h-1 at 30 bar and 18 489 g/g Cr h-1 at 40 bar. Even though this decrease in activity was observed, it was interesting to note that this catalyst was still more active than 1, the SNS(ethyl) homogeneous catalyst. This decrease can be attributed to a combination of two factors, viz. the solubility of catalyst 2 and the enhanced activity by support interaction.
As was the case with catalyst 1, catalyst 2 at both pressures maintained comparable selectivities to both C6 and 1-C6.At30 bar ethylene, catalyst 2 achieved 98.2 and 94.3 % selectivity to C6 and 1-C6 (defined as the % of 1-C6 in the C6 fraction), respectively. At 40 bar, the selectivities that the same catalyst achieved were 97.3 and 90.9 % to C6 and 1-C6, respectively. Catalyst SC2 formed less PE than 2. A similar trend regarding the make-up of the C6 fraction was observed between catalysts 2 and SC2. Selectivities to 1-C6 within the C6 fraction of 44.3 and 45.2wt % were achieved, and the highest of these was achieved at 40 bar with SC2. The pattern of these catalysts with regard to 1-C6 selectivity is that it increases with pressure for both catalysts 2 and SC2. However, this is not the case with catalyst activity, as for SC2, it was observed that it decreased with the increase in ethylene pressure. The catalytic performance of SL2 was the lowest of the three decyl substituted catalysts, achieving 2199 g/g Cr h-1 at 30 bar and 2572 g/g Cr h-1 at 40 bar. The overall C6 selectivity also decreased. The PE formed was twice as much as that over the catalyst which is structurally similar to it, i.e. SL1. The support effect in catalysts SC2 and SL2 is similar to that in SC1 and SL1, causing decreased 1-C6 selectivities. Catalyst activity of SC2, however, decreased with increased pressure. In this regard, comparing catalysts 2 and SC2 can be likened to 1 and SL1. The bulkiness of the support dominates over the stability it offers, likely due to steric interaction with a much bulkier alkyl chain.
3.2.2.2. Cr-PPP Catalysts
Chromium complexes of tridentate PPP ligands, have been reported to be highly active and selective for ethylene trimerization.28 The authors noted that this class of catalysts underwent a thermally induced catalyst deactivation at higher temperatures. The Cr-PPP catalysts reported in this study were chosen to study the effect of temperature under constant pressure and investigate the support's role in aiding or abating thermal deactivation. Table 6 shows the catalytic testing results for the Cr-PPP catalysts at 30 bar ethylene pressure. With the homogeneous catalyst, 3, the highest activity obtained was 15 641 g/g Cr h-1 at 60 °C. The activity of the catalyst decreased as the temperature was increased. At the highest temperature of 90 °C, the activity of 3 decreased to 4210 g/g Cr h-1, probably due to catalyst deactivation. Catalyst deactivation/decomposition changes the catalyst properties to that of a polymerization catalyst. This decrease in activity is evident because, as the selectivity to 1-C6 decreased, more PE was formed. The 1-C6 in C6 selectivity at this temperature also decreased to 78.6 wt %.
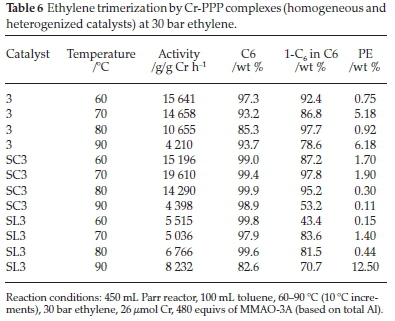
When the supported counterpart SC3 was tested, it also showed relatively high activities. The highest activity was at 70 °C giving 19 610 g/g Cr h-1. This activity was higher than any activity determined for catalyst 3 at all temperatures. While it was expected that the activity would increase further with temperature, it showed the opposite effect. At 80 °C the activity decreased to 14 290 g/g Cr h-1 and eventually to 4398 g/g Cr h-1 at 90 °C. Thus thermally induced deactivation was likely, and thus the failure of the support to abate thermal deactivation was observed. Results for catalysts 3 and SC3 suggested that the catalysts reached their maximum activity in the range 60-70 °C, with higher temperatures deactivating them. The selectivity to 1-C6 in C6 decreased to 78.6 wt% at 90 °C with catalyst 3, and significantly with SC3 to 53.2 wt %. The selectivity shifts to other C6 products.
The catalytic results of SL3, however, showed a different trend altogether. At 60 °C an activity of 5515 g/g Cr h-1 was achieved, which decreased to 5036 g/g Cr h-1 on increasing the temperature to 70 °C. Between 70-90 °C, there was an increase in the activity of SL3 to 8232 g/g Cr h-1. This trend was unexpected as catalysts 3 and SC3 showed deactivation with temperature increases. What accompanied this apparent catalyst activity improvement of SL3 with temperature increase was a decrease in selectivity to 1-C6, which would explain the increased amount of PE formed, namely 12.5 % compared to 0.15,1.4,0.44 % at 60, 70 and 80 °C, respectively.
Table 7 shows the catalytic testing results for the synthesized Cr-PPP catalysts at 40 bar ethylene pressure. The activity of catalyst 3 decreases as the temperature increase with the highest activity of 19 416 g/g Cr h-1 achieved at 60 °C. The slight increase in pressure does not seem to influence the activity trend of catalyst 3 compared to results observed at 30 bar in that it also decreased with increasing temperature. However, it can be noted that even though the catalyst activity trend with temperature is maintained between these pressures, catalyst 3 at 40 bar has higher catalyst activities. The selectivity to 1-C6 also remains comparable to the results obtained at 30 bar and at 90 °C, where the lowest catalyst activity was recorded. The selectivity to C6 decreases at 90 °C and more PE is formed than at the other temperatures. This trend is also observed with catalyst SC3. The reverse order is observed for catalyst SL3, which suggests that the interaction of the catalyst with the support is favoured, as catalyst activity seems to increase with increased temperature and improves with an increase in pressure as well.

The supported catalyst SC3 achieved the highest catalytic activity of 14 998 g/g Cr h-1 at 60 °C, which decreased with increasing temperature to 7944 g/g Cr h-1 at 90 °C. What was notable at this point was that SC3 formed more PE at 60, 70 and 90 °C than it did at the same temperatures at 30 bar. At 90 °C, the highest examined temperature, the selectivity to 1-C6 decreased significantly to 61.9 wt %, and it gave 15.2 wt % PE, giving the catalyst its poorest performance. Only 70.3 wt % of the C6 fraction was 1-C6. Catalyst SL3, on the contrary, was observed to perform best at 90 °C, achieving 12 542 g/g Cr h-1, as activity increased with temperature. The highest selectivity to 1-C6 was achieved at this temperature, giving 95.2 wt %. Compared to SC3 at 60 °C, which has the lowest activity, this suggests that the pressure and temperature do not affect and influence the differently supported catalysts in the same way. Furthermore, catalyst SC3 seems to undergo deactivation at high temperatures, which is not observed for catalyst SL3. It can be inferred from this catalyst behaviour that the mode of interaction with the support affects the kinetics of the reaction.
In terms of catalyst activity, the homogeneous catalyst 3 showed thermal instability with increased temperature. It was hoped that the support would enhance activity by inducing thermal stability as observed with SL1. It was generally not the case. The support, however, was observed to enhance catalyst activity relative to the unsupported catalyst at 90 °C, which is the apparent advantage of the support. This enhancement was at the expense of selectivity to 1-C6. The weak interaction of SL3 with the support renders the catalyst 'homogeneous-like' in terms of catalyst behaviour. The influence and presence of the support, in this case, becomes evident at higher temperatures.
4. Conclusion
The catalytic results showed that the substituent on the sulphur donor atom remains very important for the catalytic activity. The decyl substituted catalyst was the most active, both for the supported and the unsupported catalysts. The selectivity to 1-C6 within the C6 fractions formed depended on the method used to support the catalysts. The catalysts supported with the complex directly on the support (SC1 and SC2) were superior in activity, selectivity to 1-C6 and gave lower PE formation compared to SL1 and SL2. In terms of catalytic behaviour, catalysts SL1 and SL2, even though showing lower activities, followed the same trend of catalysts 1 and 2, respectively, in how they were affected by similar reaction conditions. This catalytic behaviour suggests that SL1 and SL2 behave as homogeneous catalysts in ethylene trimerization. It is suggested that this could imply a very weak interaction with the support (physisorption) and, hence, possibly leaching.
The catalyst performance of the Cr-TAC catalyst improved with supporting the metal complexes. An increase in temperature proved to favour the selectivity towards 1-C6. Even though the supported catalysts are much more active than their homogeneous counterparts, with regard to selectivity, the homogeneous catalysts are superior. The influence of increasing temperature on the Cr-PPP catalysts induces thermal deactiva-tion at both 30 and 40 bar ethylene. A role of the support in abating thermal deactivation is observed with catalyst SL3 and not SC3. Instead of the ligand, the method of supporting the metal seems to be important for improving the activity and selectivity of the catalyst. Increasing reaction temperature favoured the catalysts that were not favoured by increasing pressure, i.e. SC3, SL3 versus SC1, SC2, SL1, SL2.
Supplementary Material
Supplementary information is provided in the online supplement.
ORCID iDs
M.L.Shozi: orcid.org/0000-0001-7666-2543
X.Zulu: orcid.org/0000-0001-6951-8391
H.B. Friedrich: orcid.org/0000-0002-1329-0815
References
1 F. Yu, Y. Yang, D. He, D. Gong and Z.-R. Chen, Pressure-sensitive supported FI catalyst for the precise synthesis of uni- and bimodal polyethylene, ind. Eng. Chem. Res., 2017, 56, 4684-4689. [ Links ]
2 H. Rahiala, I. Beurroies, T. Eklund, K. Hakala, R. Gougeon, P. Trens and J. B. Rosenholm, Preparation and characterization of MCM-41 supported metallocene catalysts for olefin polymerization, J. Catal., 1999,188, 14-23. [ Links ]
3 E. Y.-X. Chen and T.J. Marks, Cocatalysts for metal-catalyzed olefin polymerization: activators, activation processes, and structure-activity relationships, Chem. Rev., 2000, 100, 1391-1434. [ Links ]
4 J.H.Z. dos Santos, A.E. Gerbase, K.C. Rodenbusch, G.P. Pires, M. Martinelli and K.M. Bichinho, Hybrid supported zirconocene and niobocene catalysts on MAO-modified silicas, J. Mol. Catal. A: Chem., 2002,184, 167-173. [ Links ]
5 M. Galan-Fereres, T. Koch, E. Hey-Hawkins and M.S. Eisen, Synthesis and olefin polymerization using supported and non-supported geometry constrained titanium complexes, J. Organomet. Chem.,1999, 580, 145-155. [ Links ]
6 H. Juvaste, E.I. Iiskola and T.T. Pakkanen, Aminosilane as a coupling agent for cyclopentadienyl ligands on silica, J. Organomet. Chem., 1999, 587, 38^5. [ Links ]
7 R.M. Kasi and E.B. Coughlin, Supported constrained-geometry catalysts on cross-linked (aminomethyl)polystyrene: studies of ethylene and 1-octene polymerizations, Organometallics, 2003, 22, 1534-1539. [ Links ]
8 J.M. Thomas, R. Raja and D.W. Lewis, Single-site heterogeneous catalysts, Angew. Chem. Int. Ed., 2005,44, 6456-6482. [ Links ]
9 F. Silveira, M. d. C.M. Alves, F.C. Stedile, S.B. Pergher, A. Rigacci and J.H.Z. d. Santos, Effect of the silica texture on the structure of supported metallocene catalysts, J. Mol. Catal. A: Chem, 2009,298,40-50. [ Links ]
10 C.N. Nenu, E. Groppo, C. Lamberti, A.M. Beale, T. Visser, A. Zecchina and B.M. Weckhuysen, Dichloromethane as a selective modifying agent to create a family of highly reactive chromium polymerization sites, Angew. Chem. Int. Ed., 2007,46, 1465-1468. [ Links ]
11 E. Blart, J.P. Genêt, M. Safi, M. Savignac and D. Sinou, Palladium(O)-catalyzed substitution of allylic substrates in an aqueous-organic medium, Tetrahedron, 1994, 50, 505-514. [ Links ]
12 W. Leitner, Recent advances in catalyst immobilization using supercritical carbon dioxide, Pure Appl. Chem., 2004, 76, 635-644. [ Links ]
13 D.A. Annis and E.N. Jacobsen, Polymer-supported chiral Co(salen) complexes: synthetic applications and mechanistic investigations in the hydrolytic kinetic resolution of terminal epoxides, J. Am. Chem. Soc., 1999,121, 4147-4154. [ Links ]
14 J.M. Fraile, J.I. Garcia, J. Massam and J.A. Mayoral, Clay-supported non-chiral and chiral Mn(salen) complexes as catalysts for olefin epoxidation, J. Mol. Catal. A: Chem, 1998,136, 47-57. [ Links ]
15 M.R. Maurya, S.J.J. Titinchi, S. Chand and I.M. Mishra, Zeolite-encapsulated Cr(III), Fe(III), Ni(II), Zn(II) and Bi(III) salpn complexes as catalysts for the decomposition of H2O2 and oxidation of phenol, J. Mol. Catal. A: Chem., 2002,180, 201-209. [ Links ]
16 A.J. Sandee, D. Dimitrijevic, R.J. van Haaren, J.N.H. Reek, P.C.J. Kamer and P.W.N.M. van Leeuwen, Silica immobilised palladium phosphine complexes as recyclable, regioselective catalysts for the allylic alkylation, J. Mol. Catal. A: Chem, 2002,182 183, 309-317. [ Links ]
17 H. Zhang, S. Xiangand C. Li, Enantioselective epoxidation of unfunc-tionalised olefins catalyzed by Mn(salen) complexes immobilized in porous materials via phenyl sulfonic group, Chem. Commun., 2005, 1209-1211. DOI: 10.1039/B417041E [ Links ]
18 R. Chen, R. P.J. Bronger, P.C.J. Kamer, P.W.N.M. van Leeuwen and J.N.H. Reek, Noncovalent anchoring of homogeneous catalysts to silica supports with well-defined binding sites, J. Am. Chem. Soc., 2004, 126, 14557-14566. [ Links ]
19 P.W.N.M. van Leeuwen, A.J. Sandee, J.N.H. Reek and P.C.J. Kamer, Xantphos-based, silica-supported, selective, and recyclable hydro-formylation catalysts: a review, J. Mol. Catal. A: Chem, 2002,182-183, 107-123. [ Links ]
20 K.A. Alferov, G.P. Belov and Y. Meng, Chromium catalysts for selective ethylene oligomerization to 1-hexene and 1-octene: recent results, Appl. Catal. A: General, 2017, 542, 71-124. [ Links ]
21 S. Sahoo, P. Kumar, F. Lefebvre and S.B. Halligudi, Oxidative kinetic resolution of alcohols using chiral Mn-salen complex immobilized onto ionic liquid modified silica, Appl. Catal. A: General, 2009, 354, 17-25. [ Links ]
22 R. Guimaräes, F. C. Stedile and J.H.Z. dos Santos, Ethylene polymerization with catalyst systems based on supported metallocenes with varying steric hindrance, J. Mol. Catal. A: Chem, 2003, 206, 353-362. [ Links ]
23 J.R. Severn, J.C. Chadwick, R. Duchateau and N. Friederichs, "Bound but not gagged" immobilizing single-site α-olefin polymerization catalysts, Chem. Rev, 2005,105, 40734147. [ Links ]
24 M. Fallahi, E. Ahmadi, A. Ramazani and Z. Mohamadnia, Trimeri-zation of ethylene catalyzed by Cr-based catalyst immobilized on the supported ionic liquid phase, J. Organomet. Chem.,2017,848,149-158. [ Links ]
25 T. Monoi and Y. Sasaki, Silica-supported Cr[N(SiMe3)2]3/isobutyl-alumoxane catalyst for selective ethylene trimerization, J. Mol. Catal. A: Chem., 2002,187, 135-141. [ Links ]
26 C.N. Nenu and B.M. Weckhuysen, Single-site heterogeneous Cr-based catalyst for the selective trimerisation of ethylene, Chem. Commun, 2005, 1865-1867. DOI: 10.1039/B417938M [ Links ]
27 D.S. McGuinness, P. Wasserscheid, W. Keim, D. Morgan, J.T. Dixon, A. Bollmann, H. Maumela, F. Hess and U. Englert, First Cr(III)-SNS complexes and their use as highly efficient catalysts for the trimerization of ethylene to 1-hexene, J. Am. Chem. Soc, 2003,125, 5272-5273. [ Links ]
28 D.S. McGuinness, P. Wasserscheid, W. Keim, C. Hu, U. Englert, J.T. Dixon and C. Grove, Novel Cr-PNP complexes as catalysts for the trimerisation of ethylene, Chem. Commun., 2003, 334-335. DOI: 10.1039/B210878J [ Links ]
29 A.A. Danopoulos, A.R. Wills and P.G. Edwards, The synthesis of tridentate dialkylamino ligands containing tertiary phosphorus or arsenic donors, Polyhedron, 1990, 9, 2413-2418. [ Links ]
30 J.C. Fierro-Gonzalez, S. Kuba, Y. Hao and B.C. Gates, Oxide- and zeolite-supported molecular metal complexes and clusters: physical characterization and determination of structure, bonding, and metal oxidation state, J. Phys. Chem. B, 2006, 110, 13326-13351. [ Links ]
31 T. Takei, K. Kato, A. Meguro and M. Chikazawa, Infrared spectra of geminal and novel triple hydroxyl groups on silica surface, Colloids Surf. A: Physicochem. Eng. Aspects, 1999,150, 77-84. [ Links ]
32 C.N. Nenu, P. Bodart and B.M. Weckhuysen, Turning a Cr-based heterogeneous ethylene polymerisation catalyst into a selective ethylene trimerisation catalyst, J. Mol. Catal. A: Chem.,2007,269,5-11. [ Links ]
Received 12 February 2020
Revised 11 May 2020
Accepted 12 July 2020
* To whom correspondence should be addressed. E-mail: friedric@ukzn.ac.za
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]

