










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.75 Durban 2021
http://dx.doi.org/10.17159/0379-4350/2021/v75a3
RESEARCH ARTICLE
https://doi.org/10.17159/0379-4350/2021/v75a3
Quantum Chemistry of Cocaine and its Isomers I: Energetics, Reactivity and Solvation
Safa Ben AmaraI; Thorsten KoslowskiII; Ali ZaidiI, *
ILabomtoire de Spectroscopie Atomique Moleculaire et Applications, Université de Tunis El Manar Faculté des Sciences de Tunis, Campus Universitaire El-Manar, 2092 El Manar, Tunis, Tunisia
IIInstitut für Physikalische Chemie, Universität Freiburg, Albertstraße 21, 79104 Freiburg im Breisgau, Germany
ABSTRACT
We investigate the rich stereochemistry of cocaine and its diastereoisomers from a theoretical perspective using density functional theory. The relative stability of the eight considered isomers is discussed, and a comparison of the corresponding internal coordinates is given. Our results reveal that the S-pseudococaine isomer is the most stable conformation, whereas the natural occurring isomer (R-cocaine) lies higher in energy. The different isomers' chemical reactivity is discussed based on the calculation of the hardness, softness, electrophilicity and dipole moment. It was found that the dipole moment varies over a broad range from 0.65 to 4.60 D, whereas the other properties are slightly modified. The solvent effect on the energy stability of the cocaine isomers was studied by considering chloroform, dimethyl-sulfoxide (DMSO) and water as implicit solvents. Our calculations show that the different isomers' energy order and their energy gaps are slightly modified due to solvent effects. However, in all cases, the S-pseudococaine remains the most stable isomer. However, the dipole moment and the chemical reactivity of the cocaine isomers increase with the solvent polarity.
Keywords: Cocaine isomers,DFT, stability, solvent effect, chemical reactivity.
1. Introduction
One hundred forty-five years after discovering and rationalizing stereochemistry by Van't Hoff and le Bel, it remains a fascinating subject in chemistry and biology. The topic is particularly interesting in biochemistry and medicinal chemistry, as stereoisomers of the same molecule may have different physiological effects.1-3 Apart from the interaction with polarized light, enantiomers with a single chiral atom show identical physical properties. On the other hand, diastereomers with two or more chiral centres differ in their geometry and energetics. A detailed analysis of the structure and changes in geometrical and energetic parameters would enable us to understand many biological molecules' chemical properties and their interaction with other compounds in their metabolism.4-7 Some diastereoisomers may not be accessible experimentally, either due to the lack of synthetic paths or due to their instability. In these cases, information can be obtained theoretically from the knowledge of the corresponding stereoisomers' relative stabilities and their internal conversion.
In some particular cases, it becomes primordial to acquire a complete description of the chemical properties of the different isomers of a given compound. For example, aspects of cocaine stereochemistry have even become a legal issue.8 It also has implications on drug analytics, where it is important to demonstrate that a sample contains the naturally occurring l-cocaine.9
The l-cocaine isomer is extracted from leaves of the coca plant. Despite some medicinal benefits, it is considered one of the most dangerous and addictive drugs, and thus, it was subjected to extensive chemical studies. As shown in Fig. 1, the cocaine molecule (benzoylmethylecgonine) has the formula 2-(methoxycarbonyl)-3-(benzoyloxy)tropane. Its stereochemistry is based on the following principles:
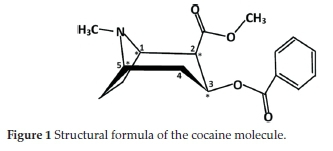
• The tropane ring can exist in four conformers: the six-membered ring can form either a chair or a boat conformation, and for both cases, the N-methyl group could be in an axial or an equatorial position. These stereochemical behaviours have been studied theoretically or experimentally by X-ray crystallography.10-14 The chair conformation of the tropane ring with the N-methyl group in an equatorial position is found to be the preferred one.
• The four chiral carbon atoms, named C1, C2, C3 and C5, lead to 16 different stereoisomer combinations. However, stable isomers must have C1 and C5 in opposite R-S configurations. Consequently, only eight stereoisomers are considered: four diastereoisomers are known as R-cocaine (or l-cocaine), R-pseudococaine, R-allococaine and R-allopseudococaine and their corresponding enantiomers S-cocaine, S-pseudococaine, S-allococaine and S-allopseudococaine.
The synthesis and elucidation of the molecular structure of cocaine isomers were performed by Willstätter et al.15These authors succeeded to synthesize cocaine and pseudococaine, but the two remaining cocaine isomers (allococaine and allopseudococaine) had not been isolated. Several decades later, Findlay16 and Hardegger and Ott11 used classical chemical methods to investigate the three-dimensional structure of cocaine and pseudococaine, and in later works Findlay17-18 succeeded to establish the structure of the allococaine and allopseudococaine isomers. The molecular structure of the cocaine isomers has been endorsed using NMR spectroscopy12,19 and a conformation study of the piperidine ring of the tropane nucleus has been performed.12 For the natural occurring R-cocaine, a molecular structure based on an X-ray diffraction study has been put forward by Gabe and Barnes.14 The optical differentiation of the R-cocaine and S-cocaine isomers was suggested by Eskes20 using a thin-layer chromatographic procedure. The separation of the cocaine, pseudococaine, allococaine and allopseudococaine isomers has been described using the ion-pair reverse-phase high-performance liquid chromatography.21 An early attempt, to describe the cocaine geometry and its energetics, has been performed by Villar and Loew using molecular mechanics and semi-empirical calculations.10 Recently, Fagan el al. have combined chiroptical methods and DFT calculations to access the structure of cocaine in solution.22
Due to the importance of the stereochemistry of cocaine, we aim in the present work to give a complete description of the eight possible stereoisomers of the cocaine molecule using theoretical methods. Discussion about the relative stability of these isomers will also be presented.
On the other hand, the disparate biological response of drugs also motivates an investigation of their reactivity to distinguish between the active and the inactive stereoisomers. Herein, global quantities such as electric dipole moment (μ), hardness (η), electronegativity (χ) and electrophilicity (ω) have been calculated and considered as reactivity indices.
Since the cocaine compounds are naturally found as a powder and the cocaine metabolism occurs in solution, additional study taking into account the solute-solvent interactions was undertaken. The solvent's effect on the relative energy stability of cocaine isomers was studied by considering organic (chloroform and DMSO) and inorganic (water) solvents. These solvents were chosen according to the following criteria: chloroform is an aprotic low polar solvent with a small dielectric constant (ε = 4.8), DMSO is an aprotic polar solvent with a higher dielectric constant (ε = 47.2), while water is a protic high polar solvent with a large dielectric constant (ε = 79).
We also consider these computations as a case study, illustrating how theoretical methods can help address stereochemistry problems. They form the second and final part of our work's geometrical basis, which will address these model compounds' theoretical infrared and NMR spectroscopy.
Hence, the remaining part of this work is organized as follows: we briefly review the methods applied here, discuss the geometry and its relation to experiment, investigate the question of the stability of the isomers, discuss their chemical reactivity and in a final step, comment on their behaviour in solution, as modelled by a reaction field.
2. Computational Methods
The molecular structure of the ground electronic state of the different cocaine isomers (Fig. 2) was fully optimized, within the framework of density functional theory (DFT) as implemented in the Gaussian 16 program.23 This optimization was done in combination with the standard split-valence basis set with added diffuse and polarization functions 6-311 + +g(d,p).24,25 This basis set is considered sufficient while doing DFT calculations.
The structural parameters were calculated at a Hartree-Fock (HF) starting level. These results were improved using the Becke three-parameter exact exchange functional (B3)26 combined with the gradient corrected correlation functional of Lee-Yang-Parr (LYP) leading to the commonly used B3LYP hybrid func-tional.27 For the obtained geometries, the frequencies were determined from the second derivatives of the energy computed using analytically calculated first derivatives to establish the stationary points. All optimized structures were checked by analysis of harmonic vibration frequencies. Positive values of all the calculated vibrational wavenumbers and the presence of six zero-frequency modes confirmed the geometry to be located at the true local minima on the potential energy surface.
Within the DFT theoretical chemistry tool, the molecules' ground state's molecular properties could be derived from the frontier molecular orbital (FMO) theory. Quantitative definitions of the electronegativity (χ) and the hardness (η)28 correspond to the first and the second derivatives of the total electronic energy with respect to the number N of electrons at fixed external potential 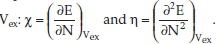
Using the usual finite difference formula electronegativity and hardness are given by: χ =  (IP + EA) and η = (IP - EA) where IP is the ionization potential, and EA is the electron affinity.
(IP + EA) and η = (IP - EA) where IP is the ionization potential, and EA is the electron affinity.
For closed-shell molecules, the molecular orbital theory based on the Koopmanns' theorem enables us to relate the IP to the HOMO (high occupied molecule orbital) energy and EA is obtained as EA = -ELUMO, where LUMO refers to low unoccupied molecule orbital.
So the hardness could be expressed by η = ELUMO -EHOMO, and 1
the electronegativity becomes 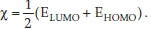 (Elumo + Ehomo) .
(Elumo + Ehomo) .
The global electrophilicity index given by:29  is a powerful tool for studying the chemical molecule reactivity, because it contains the electrophilic effect, described by χ, and the nucleophilic effect, described by η. So, a high χ value and a low η value leads to the more electrophilic compound.
is a powerful tool for studying the chemical molecule reactivity, because it contains the electrophilic effect, described by χ, and the nucleophilic effect, described by η. So, a high χ value and a low η value leads to the more electrophilic compound.
We have also calculated the electric dipole moment (μ) in gas and solution phases for the different isomers. It is an important molecular property, which describes the polarity of the molecule, and it is particularly useful for the discussion of the solute-solvent interactions.
3. Results and Discussion
3.1. Molecular Geometry Optimization and Relative Stability
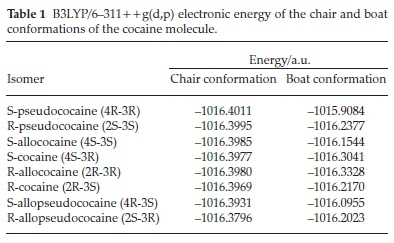
As a preliminary step, we carried out a DFT calculation using the 6-311 + +g(d,p) basis set for the eight stereoisomers of cocaine taking into account the two possible chair and boat conformation of the tropane ring.
The obtained results are summarized in Table 1, where we can remark that the chair conformation is more stable for all studied isomers than the boat conformation. This result is consistent with the preferential chair conformation obtained experimentally for all isomers.12,14,19 In the following studies, we will focus our attention on the cocaine isomers with chair conformation.
The eight stereoisomers' optimized structure of the title molecule at the B3LYP/6-311 + +g(d,p) are displayed in Fig. 2. According to their stereochemistry relationship, these compounds are presented as four enantiomeric groups: R-S cocaine, R-S pseudococaine, R-S allococaine and R-S allopseudococaine. The numbering schemes of atoms in the studied cocaine isomers and the R-S configurations of the chiral carbon atoms are also given.
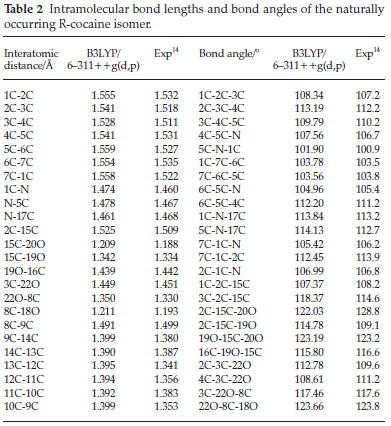
Table 2 summarizes the geometrical parameters of the naturally occurring R-cocaine isomer, and for the other isomers, geometries are available on request. As we can see from Table 2 calculations lead to a good accuracy with regard to experimental results14 for both bond and angle geometrical parameters. Our calculations well reproduce the bond distances since they correspond to a maximum discrepancy of a 0.04 A with the experimental values. This deviation from the experimental results is found for C-C bond lengths of the phenyl moiety. Our results look quite satisfactory with regard to bond angles since the maximum error is 4 °. Indeed, the calculated internal coordinates for the other isomers reflect a small change in distances when moving from any isomer to another and they could be sent on request.
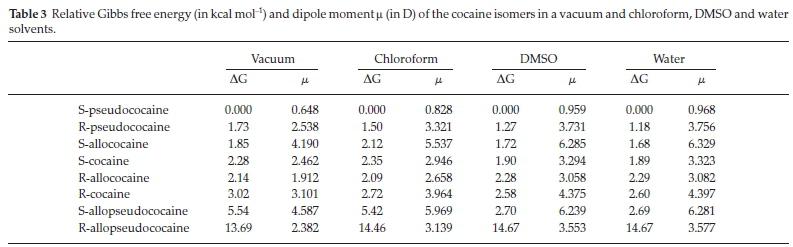
In order to discuss the relative stability of the different stereoisomers of the cocaine molecule in the gas phase, we give in Table 3 the Gibbs free energy gaps ∆G at 298.15 K of the studied stereoisomers. The most energetically favourable stereoisomer was found to be the S-pseudococaine. By contrast, the naturally occurring R-cocaine isomer lies 3 kcal mol-1 higher in energy than the S-pseudococaine compound. This 'unusual' behaviour could be explained by the fact that our study is done in the gas phase. Whereas natural cocaine is obtained in the condensed phase, a further stabilization would occur by interaction with the natural medium's chemical environment.
In Fig. 3, we display a relative energy plot where we can see that the most stable isomer is the S-pseudococaine and the least stable one is the R-allopseudococaine. For each enantiomeric pair, we found that S isomer is more stable than its R enantiomer. For the most stable pseudococaine isomers, the R-enantiomer (2) is less stable than the S-enantiomer (1) by 1.73 kcal mol-1. For the enantiomeric allococaine pair, the S enantiomer (3) is more stable than the R enantiomer (4) by 0.3 kcal mol-1. For the cocaine enantiomeric pair, we found that S-cocaine (5) is more stable than R-cocaine (6) by 0,7 kcal mol-1. The allopseudococaine enantiomeric pair exhibits the same energetic order with the S-enantiomer (7) more stable than the R-isomer (8). However, the obtained energy gap of 8.43 kcal mol-1, in this case, is larger than the previous energy gaps calculated for the three other enantiomeric groups.
Since the different isomers of the cocaine molecule correspond to a change in the methoxycarbonyl position from C2 to C4 and the switch between α and β positions of the methoxycarbonyl and the benzoyloxy groups, we can establish a relationship between these isomers and discuss their relative stability. Based on the different isomers' equilibrium structures displayed in Fig. 2, the corresponding changes are summarized in Fig. 4. From Figs. 2-4, we can expect that relative thermodynamic stability of the cocaine isomers seems to be related to steric demand. The most stable cocaine isomers (pseudococaine and allococaine) have methoxycarbonyl and benzoyloxy groups in opposite α and β positions, whereas the less stable isomers (cocaine and allopseudococaine) have the two groups with the same α or β positions. In particular, R-allopseudococaine is the least stable isomer. It lies substantially high in energy due to steric hindrance involving the methoxycarbonyl group, the benzoyloxy group and the tropane ring.
3.2. Global Reactivity Indicators
Understanding reactivity is one of the challenging chemistry tasks, aiming to elucidate the brain's drug action mechanism. In the particular case of cocaine, there have been significant advances in understanding the mechanism of its action on the central nervous system. It was found that cocaine inhibits the dopamine transporter, and this action is stereoselective since only the naturally occurring R-cocaine isomer is active. By contrast, the other isomers have a less potent effect on the nervous system.
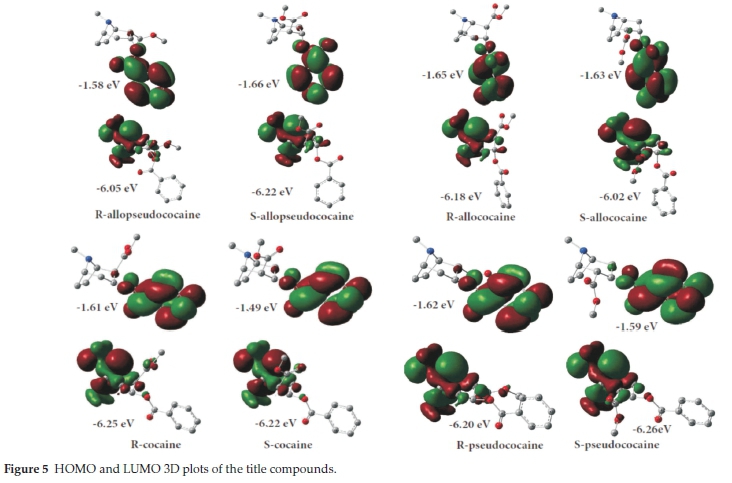
It is well established30-38 that a given molecule's kinetic stability and chemical reactivity is mainly related to the energy gap between the frontier molecular orbitals, known as HOMO and LUMO orbitals. Fig. 5 gives the 3D mapped isodensity surface plots of the HOMO and LUMO orbitals for the different cocaine isomers. According to this figure, the HOMO is mainly localized over the tropane ring, and it is of π bonding character. By contrast, the LUMO is localized on the benzoyloxy group ring, and it is of mostly π-antibonding type orbitals. In electron transfer study, the HOMO orbital acts as an electron donor and the LUMO orbital behaves as an electron acceptor.
Furthermore, the interaction strength pattern of the cocaine isomers could be investigated using the hardness (η), the electronegativity (χ) and the electrophilicity (ω) global reactivity indicators.
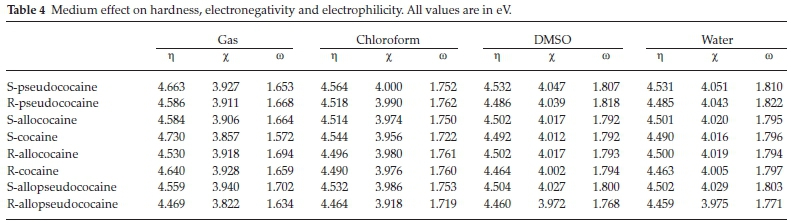
A hard molecule has a large HOMO-LUMO energy gap, showing high chemical stability. A soft molecule has a small HOMO-LUMO energy gap, which means small excitation energies to the manifold of excited states. Based on the values obtained at the B3LYP/6-311 + +g(d,p) level of theory in vacuum for the HOMO and the LUMO energies (Fig. 5), the hardness (η), electronegativity (χ) and electrophilicity index (ω) for the various cocaine compounds were calculated and tabulated in Table 4. These results show that S-cocaine is the hardest isomer with ηιηίκ = 4.730 eV whereas R-allopseudococaine is the softest isomer with = 4.469 eV. These results could be related to the LUMO destabilization in the S-cocaine and the HOMO destabilization for the R-allopseudococaine (Fig. 5).
Table 4 shows that S-allopseudococaine has the highest electronegativity value (3.940 eV) due to a LUMO stabilization, while the R-allopseudococaine isomer has the lowest electro-negativity value (3.822 eV) due to a LUMO destabilization. Overall, one can remark that for each enantiomeric pair the S-isomer has a higher hardness than the R-isomer. Still, change in hardness and electronegativity over the studied cocaine isomers is considered small and hence, their chemical reactivity would be slightly different in the gas phase.
Furthermore, we can consider a challenging chemistry index, namely the electrophilicity (ω) which is a quantitative factor that describes the energy stabilization of a chemical system when it acquires an additional electronic charge from the external environment. A high electrophilicity (ω) value indicates good electrophile character, while a lower value of electrophilicity (ω) value indicates a less electrophile character. The S-allopseudo-cocaine has the highest electrophilicity (ω), whereas the S-cocaine is the least electrophile cocaine isomer (Table 4). Based on the electrophilicity scale, organic molecules have been classified as strong electrophiles (ω > 1.5 eV), moderate electrophiles (0.8 < ω < 1.5 eV) and marginal electrophiles (ω < 0.8 eV).39 Accordingly, the cocaine isomers could be considered strong electrophiles since their electrophilicity ranges from 1.572 to 1.702 eV
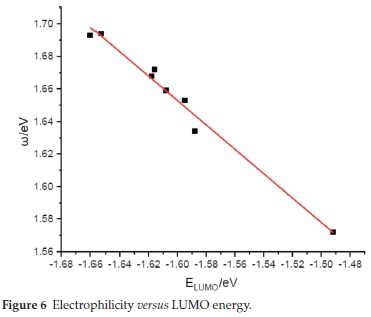
Figure 6 shows the global electrophilicity ω versus the LUMO orbital energy for the different cocaine isomers. This figure shows an excellent linear correlation relationship (y = -0.750x + 0.451) between electrophilicity ω and LUMO energy for all studied isomers with the correlation coefficient R2 = 0.98. This result is well consistent with the corresponding chemical meaning of electrophilicity ω and previously given LUMO energy, which expresses that both ω and ELUMO = -EA is related to a molecule's capability to accept an electron. Such a linear relationship was found in some previous work.40-42
Additionally, the chemical reactivity of a given molecule and its interaction with a polar medium could be investigated using its electric dipole moment. Thus, in Table 3 we also give the dipole moment μ of the ground state in the gas phase of the different cocaine isomers using B3LYP method with the 6-311 + +g(d,p) basis set. We can see that all cocaine stereo-isomers are polar, and the highest value of the dipole moment is obtained for the S-allopseudococaine isomer (μ = 4.587D). By contrast, the most stable S-peudococaine isomer has the smallest dipole moment (μ = 0.648D).
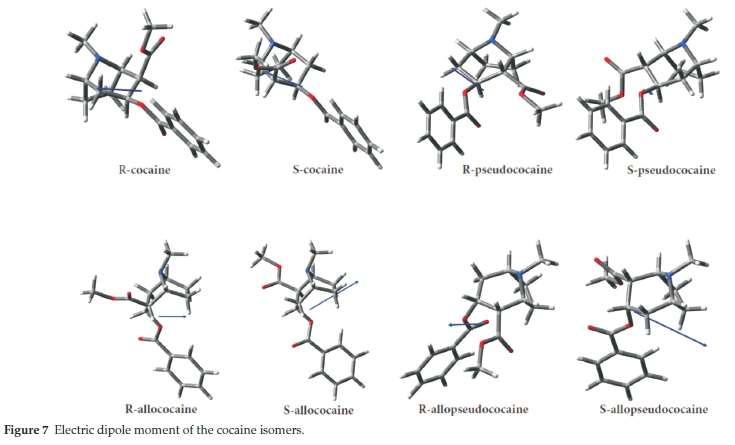
For the pseudococaine, allococaine and allopseudococaine enantiomeric pairs, there is a large difference (around 2D) in the dipole moment between each of the R and S stereoisomers. However, in the cocaine enantiomeric pair, a small difference of 0.7D is found between the R and S stereoisomers' dipole moment. This remarkable spread of dipole moment cannot be without influence on the diastereomers interacting with their environment, such as solvents, bio-membranes, binding to receptors or physicochemical properties, such as the tertiary amine pKa value. The dipole moment magnitudes can be related to the relative position or orientation of methoxyloxy and benzoyloxy groups to the N-methyl substituent.
Moreover, the electric dipole moment is a vector characterized by its magnitude and orientation, an important parameter in many chemical processes.43-47 The dipole moment vectors for title compounds have been obtained theoretically at the B3LYP method with the 6-311 + +g(d,p) and displayed in Fig. 7. One can see that for cocaine enantiomeric group there is a small change in magnitude and orientation of the dipole moment while for the allococaine pair, a larger change of both magnitude and orientation of the dipole moment is found. However, for the pseudococaine and allopseudococaine enantiomeric pairs, a notable change in magnitude is found and, interestingly, the dipole moment orientation rotates by about 180 °.
3.3 Solvent Effects
In this section, the relative stability and chemical reactivity of cocaine isomers are investigated in solution phase by considering solvents with varying dielectric constants. They are inspired by classical solvents used in a chemical laboratory, such as chloroform (ε = 4.8), DMSO (ε = 47) and water (ε = 80). Based on the polarizable continuum model (PCM), the solvent effect on the electronic structure of the title isomers was explored. For each isomer, equilibrium energy, electric dipole moment, hardness, electronegativity and electrophilicity was calculated using the B3LYP method and the 6-311 + +g(d,p) basis set.
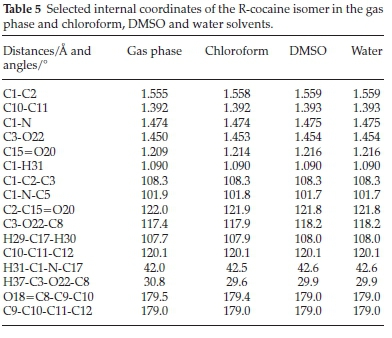
The optimized geometrical parameters for the cocaine isomers, namely bond lengths, bond angles and dihedral angles were calculated at the same level of theory as previously described. For the naturally occurring R-cocaine, we give in Table 5 selected parameters calculated in the gas phase and the presence of chloroform, DMSO and water solvents. Comparing the given internal coordinates shows that solvation had no noticeable effect on bond lengths and bond angles involving carbon or hydrogen atoms, but for C-O or C-N distances, small changes were found.
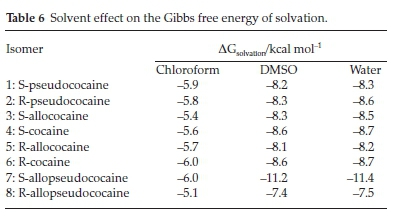
Moreover, the solvent effect on the thermodynamic stability has been investigated, and the obtained relative energies of cocaine isomers in solution are collected in Table 3. These results show that S-pseudococaine remains the most stable isomer in gas and solution phases. In the low polar chloroform solvent, the energetic order is still the same as previously found in vacuum. However, the different isomers' energy gaps are slightly larger in a solution for the S isomers. By contrast, two opposite behaviours are found for the R-isomers: R-pseudo-cocaine and R-cocaine are stabilized, but the R-allococaine and R-allopseudococaine isomers are destabilized. The largest increase of energy is obtained for R-allopseudococaine, where the relative Gibbs free energy gap becomes 14.46 kcal mol-1 in chloroform instead of 13.69 kcal mol-1 in the gas phase. However, in the considered polar solvents (DMSO and water) and apart from R-allococaine and R-allopseudococaine isomers, all the remaining compounds are stabilized in solution. Curiously, the S-allopseudococaine isomer is quite affected by the DMSO and water presence, since it is stabilized by more than 2.7 kcal mol-1 when moving from gas phase to solvent medium. Indeed, aprotic (DMSO) and protic (water) solvents give rise to similar effects since the implicit continuum solvation model does not explicitly consider solvent-solute hydrogen bond interactions.
For a further discussion of the solvent effect on the stability of the cocaine isomers, it is useful to consider a widely used quantity, namely the electric dipole moment. In the last decades, the solvent's significant effect on the dipole moment has been reported in the literature.48-51 It is found that the dipole moment is bigger in solution than in the gas phase. The dipole moment increases when going from lower to higher dielectric constant, i.e. with the solvent polarity.
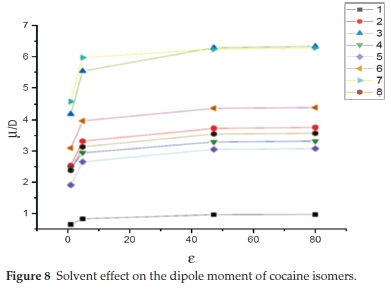
In the present work, the solvent effect on the dipole moment of cocaine isomers is shown in Table 3. The computed values of the dipole moments in the gas phase, chloroform, DMSO and water solvents are collected. These results are displayed in Fig. 8. Our results show a substantial increase in the dipole moment of cocaine isomers when going from the gas phase to solution and thus a gradual increase with increasing the solvent's polarity. In particular, the dipole moment of the S-allococaine and the S-allopseudococaine isomers have the highest values (around 6.3D) in DMSO and water with an increase of 2D relative to the gas phase values. The S-allopseudococaine isomer exhibits the highest dipole moment either in vacuum or in solution. This result could explain the energy stabilization previously found due to its electrostatic interaction with DMSO and water. On the other hand, solvents slightly affect the dipole moment of the most stable S-pseudococaine isomer, and its magnitude remains less than 1D. The computed dipole moment of the remaining R-pseudococaine, S-cocaine, R-cocaine and R-allococaine isomers increases gradually, with an average change of 1 D, with increasing polarity of the solvent.
Furthermore, the solvent effect on the chemical reactivity of the title compounds was studied using the computed electro-negativity, hardness and electrophilicity global indices. The obtained values are also given in Table 4. These results show that opposite change in electronegativity and hardness occurs when going from vacuum to solution phase. The electronegativity exhibits the same behaviour as presented by the dipole moment, i.e. an increase with the dielectric constant. The least stable R-allopseudococaine isomer has the smallest hardness and electronegativity in all solvents. Also, Table 4 shows that the electrophilicity of the different compounds increases with increasing the solvent polarity. The more electrophile isomer in water becomes the R-pseudococaine instead of the S-allo-pseudococaine isomer in a vacuum.
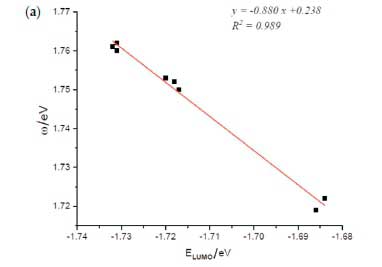
The medium effect on the linear electrophilicity-LUMO energy relationship, previously found in a vacuum has been studied in chloroform, DMSO and water. Thus, the corresponding electrophilicity versus LUMO orbital energy plots are displayed in Fig. 9a, Fig. 9b and Fig. 9c, respectively. These figures show an excellent linear correlation relationship between electrophilicity (ω) and LUMO energy in the three solvents' cases since the correlation coefficient is very close to unity (R2 ~ 0.99).


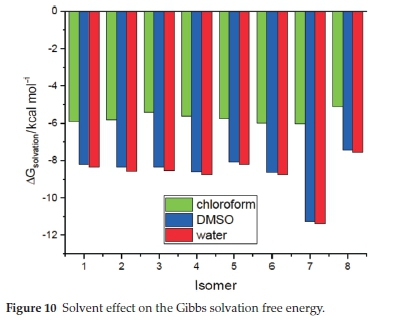
The Gibbs free energy of solvation of the title compounds was also computed, and the corresponding results are gathered in Table 6 and displayed in Fig. 10. All obtained values are negative, which means that the solvation is an exergonic process. This figure shows that solvation energy decreases with increasing the solvent's polarity. The larger change in Gibbs free energy of solvation is found for the S-allopseudococaine isomer, which could be related to its higher dipole moment value previously found. Furthermore, Fig. 10 and Fig. 8 show that the solvation energy and the dipole moment exhibit similar behaviour due to the solvent effect, i.e. a primarily large decrease from chloroform to DMSO and then little change from DMSO to water.
4. Conclusion
In this work, the relative stability and chemical reactivity of the eight possible cocaine isomers have been studied theoretically using a DFT method and the 6-311 + +g(d,p) basis set. The ground state's calculated geometry structure agrees very well with the experimental values for bond distances and bond angles. In the gas phase, the naturally occurring R-cocaine isomer is higher in energy than the most stable S-pseudococaine isomer. The presence of a solvent has a slight effect on the sequences of cocaine isomers ordered relative to their stability. Such a small solvent effect also occurs for geometrical parameters. However, a noticeable increase of the dipole moment and electrophilicity index, with the increase of the solvent polarity is found. At the same time, the global hardness exhibits a small decrease when solvent polarity increase.
So, according to our results, we can say that the thermodynamic stability of cocaine isomers is mainly related to steric demands. By contrast, the chemical reactivity and kinetic stability could depend on both steric and electronic effects.
Nevertheless, there is no single simple rationalization of the diastereomers' relative stability. Local dipoles can be combined to notably different global ones, but give no guide to stability. Certainly, steric demands of the bulky side groups play a role. Thus, we are faced with a level of complexity where intuitive approaches to molecular properties fail, and only solid quantum chemistry may give an answer.
Local dipoles and their interaction form the basis of vibrational spectroscopy. The large spectrum of net dipole moments observed in this study points at differences in the IR spectrum between diastereomers even using non-polarized infrared radiation. They may also contribute to a different local environment that may manifest as a chemical shift in NMR spectroscopy. The second and final part of our work will address these issues.
ORCID iDs
Τ. Koslowski: orcid.org/0000-0002-1844-2152
A. Zaidi: orcid.org/0000-0001-5786-7839
References
1 N. Chhabra, M. Aseri and D. Padmanabhan, A review of drug isomerism and its significance, Inl. J. App. Basic Med. Res., 2013, 3, 16-18. [ Links ]
2 L.A. Nguyen, H. He and C. Pham-Huy, Chiral drugs: an overview, Inl. J. Bio. Sci., 2006, 2, 85-100. [ Links ]
3 S.W. Smith, Chiral Toxicology: It's the Same thing ... Only different, Toxicol. Sci., 2009, 110(1), 4-30. [ Links ]
4 L.-L. Hu, C. Chen, T. Huang, Y.-D. Cai and K.-Chen Chou, Predicting biological functions of compounds based on chemical-chemical interactions, PLOS ONE, 2011, 6(12), e29491. [ Links ]
5 D. Gosavi, K.J. Salwe, D. Vimal, R. Gupta and J Mahatma Gandhi, Pharmacological significance of stereoisomerism, Inst. Med. Sci., 2010, 15(1), 21-26. [ Links ]
6 K. Singh, P. Shakya, A. Kumar, S. Alok, M. Kamal and S. P. Singh, Stereochemistry and its role in drug design, Inl. J. Pharm. Sci. Res., 2014, 5, 4644-4655. [ Links ]
7 J. Rice, K. Proctor, L. Lopardo, S. Evans and B. Kasprzyk-Hordern, Stereochemistry of ephedrine and its environmental significance: exposure and effects directed approach, J. Hazard. Mater., 2018, 348, 39-46. [ Links ]
8 J.G. Aston et al., The PEOPLE, Plaintiff and Respondent, Defendants and Appellants. Court of Appeal, Third District, California, Cr. 11261, December 13, 1984. [ Links ]
9 H.L. Schlesinger, Topics in the Chemistry of Cocaine, UNODC, 1985, 1, 63-78. [ Links ]
10 H.O. Villar and G.H. Loew, A conformational study of cocaine and its diastereomers, J. Comput. Chem., 1990,11, 1111-1118. [ Links ]
11 E. Hardegger and H. Ott, Kodguration des Cocains und Derivate der Ecgoninsaure, Helv. Chim. Acta., 1955, 38, 312-320. [ Links ]
12 I.F. Carroll, L.C. Coleman and A.H. Lewin, Syntheses and confor-mational analyses of isomeric cocaines: a proton and carbon-13 nuclear magnetic resonance study, J. Org. Chem., 1982, 47, 13-19. [ Links ]
13 N. Zhu, A. Harrison, M.L. Trudell and C.L. Klein-Stevens, QSAR and CoMFA study of cocaine analogs: crystal and molecular structure of (-)-cocaine hydrochloride and n-methyl-3p-(p-fluorophenyl) tropane-2p-carboxylic acid methyl ester, Struct. Chem., 1999,10(2), 91-103. [ Links ]
14 R.J. Hrynchurkic, R.J. Barton and B. Robertson, The crystal structure of free base cocaine, Can. J. Chem., 1983, 61, 481-487. [ Links ]
15 R. Willstätter and W. Muller, Ueber die Constitution des Ecgonine, Chem. Ber., 1898, 31, 1202-1214. [ Links ]
16 S. Findlay, The three-dimensional structure of cocaine, J. Am. Chem. Soc., 1953, 75(18), 4624-4625. [ Links ]
17 S. Findlay, The three-dimensional structure of the cocaines. Part I. Cocaine and pseudococaine, J. Am. Chem. Soc., 1954, 76, 2855-2862. [ Links ]
18 S. Findlay, The three-dimensional structures of the cocaines. II. Racemic allococaine and racemic allopseudococaine, J. Org. Chem., 1959, 24(10), 1540-1550. [ Links ]
19 A. Sinnema, L. Maat, A.J. Van Der Gugten and H.C. Beyerman, Configuration and conformation of all four cocaines from NMR spectra, Rec. Trav. Chim., 1968, 87, 1027-1041. [ Links ]
20 D. Eskes, Thin-layer chromatographic procedure for the differentiation of the optical isomers of cocaine, J. Chromatogr., 1978, 152, 589-591. [ Links ]
21 C. Olieman, L. Maat and H. Beyeman, Analysis of cocaine, pseudo-cocaine, allococaine and allopseudococaine by ion-pair reversephase high-performance liquid chromatography, Rec. Trav. Chim., 1979, 98, 501-502. [ Links ]
22 P. Fagan, L. Kocourková, M. Tatarkovic, F. Králík, M. Kuchaf,V Setnicka and P. Bouf, Cocaine hydrochloride structure in solution revealed by three chiroptical, ChemPhysChem, 2017, 18, 2258-2265. [ Links ]
23 M.J. Frisch et al. Gaussian 16 Revision A 03 (2016) Gaussian Inc.: Wallingford CT. [ Links ]
24 D.E. Woon and T.H. Dunning Jr., Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminium through argon, J. Chem. Phys, 1993, 98, 1358-1371. [ Links ]
25 F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005,7,3297-3305. [ Links ]
26 A.D. Beke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1998, 38, 3098-3100. [ Links ]
27 C. Lee, Y. Wang and R.G. Parr, Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B, 1988, 37, 785-789. [ Links ]
28 R.G. Parr and R.G. Pearson, Absolute hardness: companion parameter to absolute electronegativity, J. Am. Chem. Soc., 1983, 105, 7512-7516. [ Links ]
29 R.G Parr, L. Von Szentpaly and S. Liu, Electrophilicity index, J. Am. Chem. Soc., 1999, 121, 1922-1924. [ Links ]
30 R.C. Haddon and T. Fukunaga, Unified theory of the thermodynamic and kinetic criteria of aromatic character in the [4n + 2] annulenes, Tetrahedron Lett., 1980, 21, 1191-1192. [ Links ]
31 T.G. Schmalz, W.A. Seitz, D.J. Klein and G.E. Hite, Elemental carbon cages, J. Am. Chem. Soc., 1988,110, 1113-1127. [ Links ]
32 Z. Zhou, R.G. Parr and J.F. Garst, Absolute hardness as a measure of aromaticity, Tetrahedron Lett., 1988, 29, 4843-4846. [ Links ]
33 Z. Zhou and R.G. Parr, New measures of aromaticity: absolute hardness and relative hardness, J. Am. Chem. Soc., 1989, 111, 7371-7379. [ Links ]
34 Z. Zhou and R.G. Parr, Activation hardness: new index for describing the orientation of electrophilic aromatic substitution, J. Am. Chem. Soc., 1990,112, 5720-5724. [ Links ]
35 D.E. Manolopoulos, J.C. May and S.E. Down, Theoretical studies of the fullerenes: C34 to C70, Chem. Phys. Lett., 1991, 181, 105-111. [ Links ]
36 X. Liu, T.G. Schmalz and D.J. Klein, Favorable structures for higher fullerenes, Chem. Phys. Lett., 1992,188, 550-554. [ Links ]
37 R.G. Parr and Z. Zhou, Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters, Acc. Chem. Res., 1993, 26, 256-258. [ Links ]
38 J. Aihara, S. Oe, M. Yoshida and E.J. Osawa, Further test of the isolated pentagon rule: thermodynamic and kinetic stabilities of C84 fullerene isomers, Comput. Chem., 1996,17, 1387-1394. [ Links ]
39 L.R. Domingo, M.J Aurell, P. Pérez and R. Contreras, Quantitative characterisation of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions, Tetrahedron, 2002, 58, 4417-4423. [ Links ]
40 F. Núnez-Zarur, E. Arguello and R. Vivas-Reyes, Density functional study on electronic structures and reactivity in methyl-substituted chelates used in organic light-emitting diodes, Int. J. Quant. Chem., 2010, 110, 1622-1636. [ Links ]
41 C. Wang, Y. Fu, Q-X. Guo, and L. Liu, First-principles prediction of nucleophilicity parameters for p nucleophiles: implications for mechanistic origin of Mayr's equation, Chem. Eur. J., 2010, 16, 2586-2598. [ Links ]
42 L-G. Zhuo, W. Liao, and Z-X. Yu, A frontier molecular orbital theory approach to understanding the Mayr equation and to quantifying nucleophilicity and electrophilicity by using HOMO and LUMO energies, Asian J. Org. Chem., 2012,1, 336-345. [ Links ]
43 K. Koperwas, K. Adrjanowicz, A. Grzybowski and M. Paluch, The role of the dipole moment orientations in the crystallisation tendency of the van der Waals liquids - molecular dynamics simulations, Scientific Reports, 2020,10, 283-291. [ Links ]
44 A.J. Wain, H.N. L. Do, H.S. Mandal, H-B. Kraatz and F. Zhou, The Influence of molecular dipole moment on the redox-induced reorganisation of α-helical peptide self-assembled monolayers: an electrochemical SPR investigation, J. Phys. Chem. C., 2008,112(37), 14513-14519. [ Links ]
45 H.S. Mandal and H-B. Kraatz, Electron transfer across α-helical pep-tides: potential influence of molecular dynamics, Chem. Phys., 2006, 326, 246-251. [ Links ]
46 K.D. McNamara and R.W. Zoellner, The effects of substituent position and orientation on the structures and dipole moments of the cyanocyclohexanes using density functional theory calculations, Comput. Theor. Chem., 2019, 1170, article 112622. [ Links ]
47 K. Kristiansen, P. Stock, T. Baimpos, S. Raman, J.K. Harada, J.N. Israelachvili and M. Valtiner, Influence of molecular dipole orientations on long-range exponential interaction forces at hydrophobic contacts in aqueous solutions, ACS Nano, 2014, 8(10), 10870-10877. [ Links ]
48 M. Targema, N.O. Obi-Egbedi andM.D. Adeoye, Molecular structure and solvent effects on the dipole moments and polarizabilities of some aniline derivatives, Comput. Theor. Chem., 2013, 1012, 47-53. [ Links ]
49 M.F. Khan, R. Bin Rashid, M.Y. Mian, M.S. Rahman and M.A. Rashid, Effects of solvent polarity on solvation free energy, dipole moment, polarizability, hyperpolarizability and molecular properties of metronidazole, Bangl. Pharma. J., 2016,19, 9-14. [ Links ]
50 R. Ghiasi, M. Nemati and A.H. Hakimioun, Solvent effect on the structural electronic, spectra properties and first hyperpolarisability of W(CO)5L, L=(4-pyridyl methylene) malononitrile, J. Chil. Chem. Soc., 2016, 61(2), 2921-2928. [ Links ]
51 M.F. Khan, R. Bin Rashid, M.M. Rahman, M.D. Al Faruk, M.D.M. Rahman and M.A. Rashid, Effects of solvent polarity on solvation free energy, dipole moment, polarisabilty, hyperpolarisability and molecular reactivity of aspirin, Int. J. Pharm. Pharm. Sci., 2017,9(2), 217-221. [ Links ]
Received 10 July 2020
Revised 16 October 2020
Accepted 16 October 2020
* To whom correspondence should be addressed. E-mail: ali.zaidi@ipest.rnu.tn


{kind=link}
{kind=link}
{kind=link}
{kind=link}