










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.73 Durban 2020
http://dx.doi.org/10.17159/0379-4350/2020/v73a3
RESEARCH ARTICLE
C-H activation: a Critical Evaluation of a Published Method and its Application Towards Inherently Chiral Calix[4]arenes
Kevin J. Visagie; Luke Hodson; Gareth E. Arnott*
Department of Chemistry and Polymer Science, Stellenbosch University, Private Bag XI, Matieland, 7602, South Africa
ABSTRACT
C-H activation offers an intriguing access into inherently chiral calix[4]arenes, but has been little explored in the literature. In this article, we report our investigation into a published C-H activation method that uses carbamates to direct a palladium catalyzed C-H activation and subsequent reaction with N-bromosuccinimide. However, we show that this report is unfortunately flawed on a number of points. An earlier reported study revealed the more likely SEAr mechanism of the bromination reaction, which did not involve palladium catalysis. We nevertheless employed the SEAr bromination in an attempt to form inherently chiral calix[4]arenes, using a chiral (+)-menthyl carbamate as a directing group. Unfortunately, although the reaction was high yielding, the diastereomers formed were inseparable and we were unable to quantify their ratio. Subsequent removal of the chiral (+)-menthyl carbamate, returned a small positive optical rotation, suggesting that at least a level of asymmetric induction was achieved in the bromination to afford a non-racemic product.
Keywords: C-H activation, calix[4]arene, inherent chirality, bromination, diastereoselectivity.
1. Introduction
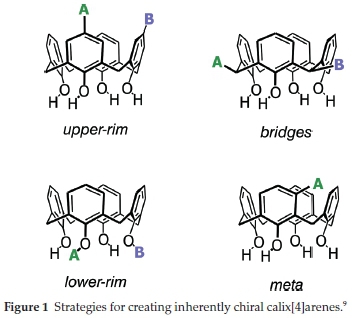
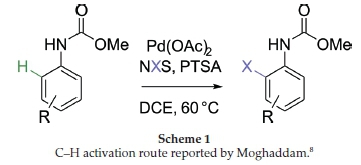
Chirality is and will always be one of the most important aspects of chemistry since all living forms are chiral. From its initial introduction to chemistry students in the form of point chirality (tetrahedral carbon atoms), students later learn that chiral molecules can be formed in many other ways. Our area of research has been focused on one of these aspects, that of inherently chiral calix[4]arenes.1,2 Inherently chiral calix[4]arenes can actually be formed by a number of different ways, which makes them attractive targets to study (see Fig. 1 for some simple examples). We have focused on meta-functionalization as a preferred strategy, owing to its similarity to planar chiral ferrocenes, which have a good history of acting as asymmetric ligands.3 To date, we have developed some strategies that stereoselectively synthesize meta-functionalized inherently chiral calix[4]arenes using ortho-lithiation chemistry directed by either chiral oxazolines4-6 or a sulfoxide.7 Whilst these methods are currently the only meaningfully stereoselective methods available to form inherently chiral calix[4]arenes, they suffer from rather difficult chemistry that makes scale-up problematic. To this end, we have been looking at other methods that might generate meta-functionalized inherently chiral calix[4]arenes. One such method involving a putative C-H activation pathway caught our attention in the literature. In this 2016 paper by Moghaddam and coworkers, it was reported that methyl carbamates were excellent directing groups for ortho-aryl C-H activation (Scheme 1).8 We wondered whether the same method could be used on a calix[4]arene to generate inherently chiral versions if the carbamate itself was chiral. Herein we would like to report our preliminary findings in this area, as well as reveal our deep concerns regarding the paper by Moghaddam and co-workers.
2. Results and Discussion
2.1. Model Study
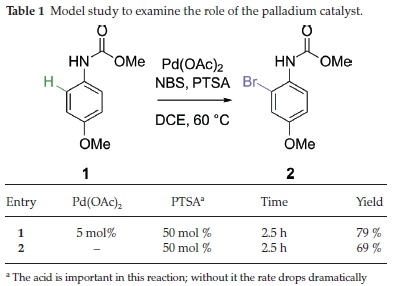
Our investigation began with a model study, primarily to ascertain whether the chemistry reported by Moghaddam and co-workers was itself correct (Scheme 1). We had some initial concerns about the reported method, as the paper appeared to have superficial errors that we found surprising. Some of these errors might have been due to topographical oversight, but did warrant further investigation. One concern was that the reported method should have been theoretically possible using just N-bromosuccinimide (NBS), without any need for the palladium catalyst. In the paper, the authors reported that the reaction failed using NBS in acetonitrile, but only worked when palladium acetate was added. The authors then reported that the reaction failed in dichloroethane (DCE) even with palladium acetate, but worked again when para-toluenesulfonic acid (PTSA) was added. The published table of results showed no example of an experiment that then excluded the palladium catalyst, but kept PTSA, i.e. a control experiment. However, the text did report that a control experiment had been performed and then referred to an incorrect entry on the table (hence a possible typographical error). For this reason, we decided to have a closer look at the reaction ourselves.
The model selected (carbamate 1) included a para-methoxy group, which served both as a model for a single functionalised aromatic ring on the calix[4]arene, and as a means for testing the directing ability of the carbamate vs. the methoxy group. Essentially, it was found that the role of the palladium in this experiment was greatly exaggerated (see Table 1), with the yield of brominated 2 only being marginally higher when it was included. In both cases, the carbamate was the sole director towards ortho-bromination. It therefore seems likely to us that Moghaddam and co-workers had somewhat overstated the importance of the palladium and its role in the reaction.
With this rather unsurprising result, we took a much closer look at the paper by Moghaddam and co-workers and noticed more problems. In the introduction, they made the main claim that: 'To the best of our knowledge, this is the first report of application of N-arylcarbamates as DG in C-halogen bond formation'. This statement cannot be proven false, since it is 'to the best of their knowledge', but it is nevertheless wrong. A quick search on Reaxsys reveals a different story: excluding papers reported after their own 2016 publication, 43 documents (including patents) report the use of NBS brominating an aryl ring ortho to a carbamate; 12 documents using NCS (chlorina-tion) and 24 documents using NIS (iodination). Many further examples can also be found employing the respective molecular halogen reagent (e.g. chlorine, bromine and iodine). A minor selection of examples from the peer review literature are shown in Fig. 2 (refer to Supplementary Information). It is disheartening that the reviewers never noticed this, since this fact alone puts a completely different interpretation onto the results presented.
Secondly, Moghaddam and co-workers cite a 2014 paper by Uhlig and Li16 making the following statement: 'Although they are structurally and electronically similar to O-aryl carbamates, after being introduced by Li et al. as an effective and removable DG, N-arylcarbamates have not been investigated as C-H activation DG(sic).' This statement is false, since Uhlig and Li very definitely reported on N-arylcarbamates being used as C-H activation directing groups. In fact, it is the entire focus of their paper, which is titled 'Aniline Carbamates: A Versatile and Removable Motif for Palladium-Catalyzed Directed C-H Activation'. In Uhlig and Li's paper, they also had a good look at the reaction mechanism and found that the aniline carbamate strongly favoured the promotion of electrophilic aromatic substitution, which aligns with the observations in the literature that NXS is itself capable of halogenating ortho to an N-aryl-carbamate via a non-C-H activation pathway.
Thirdly, closer inspection of Moghaddam and co-workers' proposed mechanism also reveals a number of problems. Firstly, an intermediate involving a deprotonated carbamate is proposed, which is unlikely since the reaction is under acidic conditions, and secondly, they make no account of the purpose of the p-toluene sulfonic acid which they point out is crucial; see ref.8: Scheme 5. A more realistic and plausible mechanism can be found in the paper by Uhlig and Li, which they cited.16 Overall, it is our opinion that Moghaddam and co-workers submitted a paper that failed to acknowledge the correct state of the art, and in so doing failed to understand the more likely interpretation of their results. The work by Uhlig and Li makes it clear that C-H activation is almost certainly occurring, but in the case of halogenation, this may not be the main mechanistic pathway. The unfortunate thing is that this paper has been cited many times, including four reviews where its conclusions have been reported without question.17-20
2.2. Calix[4]arene Study
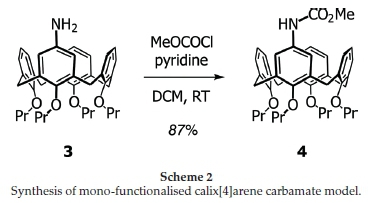
Whilst our starting point for the study proved to be somewhat spurious, the use of a chiral carbamate to potentially form inherently chiral calix[4]arenes was deemed to be worth pursuing. First an achiral model study was carried out, in order to check the chemistry, by reacting the known mono-amino-calix[4]arene 321,22with methyl chloroformate and pyridine (Scheme 2). The carbamate product 4 was confirmed via !H NMR spectroscopy (singlet at δ3.69 ppm for the hydrogen atoms of the methoxy group), infrared (1727 cm-1 for the carbamate)23 and HRMS (calculated for C42H52NO6 [M+H] + : 666.3790; found 666.3782). With this material in hand, we attempted the bromination using both Moghaddam and co-workers' method, against one that excluded the palladium catalyst. Once again, the yields of the reaction were only marginally higher when the palladium catalyst was included, suggesting that the dominant mechanism was still the electrophilic bromination and not C-H activation (see Table 2). What was also worth noting was that the unfuctionalized aromatic rings were not brominated, even though they are in principle activated by the para-propoxy groups. This is again likely due to the greater directing effect of the carbamate as evidenced in the model study (see earlier). Successful synthesis of the ortho-brominated product 5 was primarily concluded from the HRMS (calculated for C42H51BrNO6 [M+H]+: 744.2900; found 744.2872 with matchingisotopic distribution), and 1H NMR spectroscopy (whose aromatic region revealed the loss of one proton).
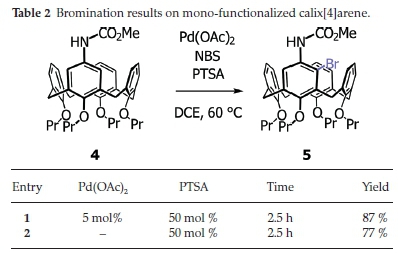
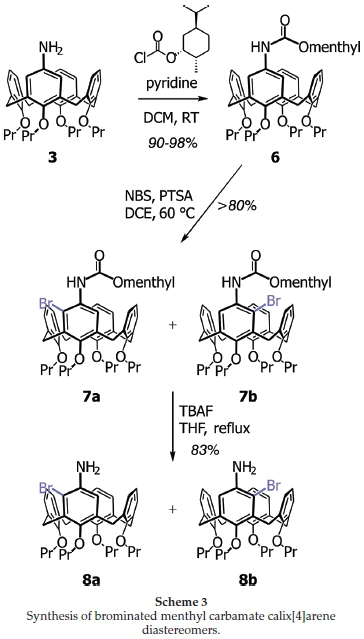
Having established that the carbamate was a suitable functional group for activating the calix[4]arene meta-position, we turned our attention to using a chiral carbamate to see whether diastereoselectivity could be achieved. For this we selected (1S)-(+)-menthyl chloroformate (it was available in our labs), and reacted it with mono-amine calix[4]arene 3 under the same conditions as before. The new chiral carbamate 6 was obtained in excellent yields between 90 and 98 % after work-up and column chromatography. The mono-menthyl carbamate calix[4]arene 6 was characterized by NMR spectroscopy, HRMS (calculated for C51H68NO6 [M+H]+: 790.5047; found 790.5040) and infrared (1697 cm-1 for carbamate).
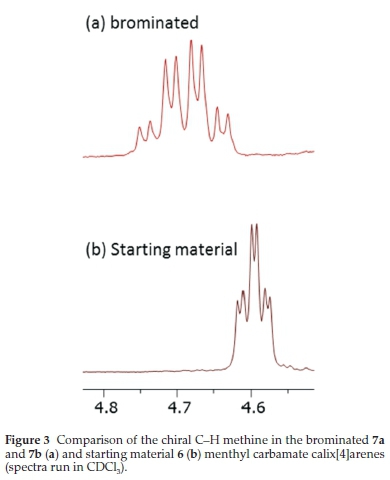
With our chiral carbamate in hand, we attempted a selective bromination reaction using the protocol without palladium, which returned a good yield (>80 %) for the inseparable brominated products 7a and 7b. We had been hoping to use 1H NMR spectroscopy to quantify the diastereoselectivity of the reaction, but to our disappointment, there were really no promising signals to work with. Different solvents (CDCl3, DMSO-d6 and C6H6) and different temperatures (up to the maximum allowed) all failed to help us determine the diastereo-selectivity. The only signal that appeared marginally useful was the methine signal on the chiral centre of the menthyl group. In the starting material this appeared at ô 4.60 ppm as a triplet of doublets, whilst in the product it appeared as a multiplet centred around δ 4.69 ppm (see Fig. 3). On close inspection, it could be seen that this multiplet was an overlay of two triplets of doublets in essentially a 1:1 ratio, suggesting a poor diastereoselectivity in the reaction.
In order to see if we could improve on this diastereoselec-tivity, we tried the reaction at lower temperatures. Since DCE is relatively limited in this sense, we changed the solvent to dichloromethane (DCM) and initiated a temperature study (Table 3). Unfortunately, in all cases (down to -35 °C) we saw no discernible improvement in the diastereoselectivity (as judged by the aforementioned signals in the 1H NMR spectra). However, what was unexpected was just how well the reaction occurred even at lower temperatures, albeit with slightly longer reaction times.
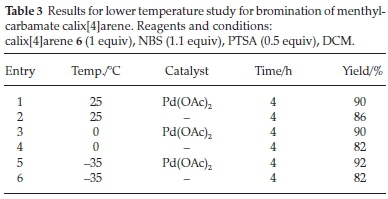
Our inability to determine the ratio of diastereomers in this reaction was frustrating. Even normal and reversed-phased HPLC experiments, including the use of a chiral column, failed to separate the two diastereomers. As a last resort, we decided to remove the chiral menthyl group and examine the optical rotation of the resultant mixture of enantiomers to see if any optical activity was displayed. Although this would not be a means of determining enantioselectivity, the optical rotation would at least point to whether any chiral induction had taken place. Removal of the menthyl group was readily achieved using Coudert's method of tetra-w-butylammonium fluoride (Bu4NF, TBAF) in THF (Scheme 3).24 The reaction, as expected,24 was sluggish and required heating under reflux for 36 h. Nevertheless, after work-up and purification, the aminocalix[4]arene product was obtained in yields >80 %. The 1H NMR spectrum showed the complete removal of the menthyl group, greatly simplifying the spectrum. The loss of the carbamate was also detected by IR spectroscopy and the HRMS returned the expected molecular ion (and isotopic distribution pattern) for the product. Optical rotation experiments were then run on material generated from bromination at -35 °C (Table 3, entries 5 and 6), returning values of [a]D = +6 ° and +3 ° for products derived from entries 5 and 6, respectively. Whilst these values cannot be used for any form of quantification, they do indicate a level of enantiomeric excess, which in turn, points to at least some degree of diastereoselectivity induced by the chiral menthyl carbamate.
3. Conclusion
In conclusion, we have shown that a report in the literature claiming a C-H activation route, mediated by a catalytic palladium in which a carbamate directs the formation of an aryl halide bond, is somewhat overstated and incorrect on a number of points claimed. Nevertheless, using a chiral calix[4]arene carbamate, bromination successfully delivered a product that suggested a modest level of inherent chirality that could not be quantified. Further work can potentially look at other chiral groups and also at extending the number of directing groups on the upper-rim of the calix[4]arene to two or even four, in order to access more interesting niete-functionalized calix[4]arenes.
Supplementary Material
Copies of NMR, IR and HRMS spectra for all new compounds synthesized and also references for halogenation reactions prior to 2016 are provided in the supplementary material appended to the end of this article.
Experimental
All chemicals were purchased from Merck or Sigma-Aldrich. Dichloromethane was dried from calcium hydride under nitrogen. Other reagents that required purification were done so according to standard procedures. The synthesis of methyl (4-methoxyphenyl)carbamate 1 was performed using a literature procedure from p-anisidine,25 and mono-aminocalix[4] arene 3 was prepared as previously reported by us.22
For syntheses performed under inert conditions the glassware was oven-dried and then placed under vacuum of <0.5 mm Hg before being periodically flushed with argon until reaching room temperature. All reactions were performed under positive pressure of 2.8 kPa of 5.0 grade argon (Air Products). Low temperature reactions were performed in a Dewar containing ice and acetone (-15 °C), solid CO2 and acetonitrile (-40 °C) or solid CO2 and acetone (-78 °C).
Column chromatography was performed using 230-400 nm silica and thin layer chromatography (TLC) was performed using Macherey-Nagel DC-Fertigfolien ALUGRAM Xtra SIL G/UV254 TLC plates. Visualization of compounds on TLC plates was performed by using a UV lamp or using a cerium ammonium molybdate (CAM) solution followed by heating.
Both 1H and 13C NMR spectra were obtained using Varian 300 MHz VNMRS, Varian 400 MHz Unity INOVA and Varian 600 MHz Unity INOVA NMR instruments. Chemical shifts were recorded using the residual solvent peaks (chloroform-d or DMS0-d6) and reported in ppm. Unless otherwise stated, NMR spectra was obtained at room temperature. All mass spectrome-try spectra were obtained by Central Analytical Facility (CAF) at Stellenbosch University using a Waters API Q-TOF Ultima mass spectrometer. IR spectra were obtained using a Thermo Nicolet Nexus FTIR instrument using the ATR attachment. Melting points were obtained using a Gallenkamp Melting Point Apparatus.
Methyl (2-bromo-4-methoxyphenyl)carbamate (2)
An oven-dried Schlenk equipped with a magnetic stir bar and flushed with argon was charged with 1 (100 mg, 0.552 mmol), NBS (108.5 mg, 1.1 eq), PTSA (48 mg, 0.5 eq) and DCE (1.1 mL) The contents were then heated to 60 °C and left to stir for 2.5 h. After 2.5 h, the reaction contents were cooled to room temperature and then diluted with DCM (20 mL). The solution was then poured into H2O (20 mL) after which the product was extracted with DCM (10 mL x 3). The organic layers were subsequently combined and washed with a 10 % HCl solution (20 mL), followed by sat. NaHCO3 (20 mL) solution and finally brine (20 mL). The solution was then dried over MgSO4 and the solvent was removed via reduced pressure. Purification was achieved via silica gel flash column chromatography (EtOAc: PET 10:90) to obtain compound 2 as an orange solid in 69 % yield (99 mg).
The characterisation data collected for this compound compared well to literature data.8
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 7.93 (br. s, 1H, NH), 7.07 (d, JHH= 2.9 Hz, 1H, ArH), 6.90-6.82 (m, 2H, ArH), 3.78 (s, 3H, OCH3) 3.77 (s, 3H, OCH3).
5-Methyl carbamate-25,26,27,28-tetrapropoxycalix[4]arene (4)
To an oven-dried 2-neck round-bottomed flask, compound 3 (375 mg, 0.62 mmol) dissolved in DCM (20 mL) and pyridine (74.0 ,uL, 0.92 mmol, 1.5 eq) was added. After cooling the mixture to 0 °C, methyl chloroformate (71.5 ,uL, 0.92 mmol, 1.5 eq) was added. The reaction was then allowed to warm to room temperature and after 30 min the reaction had run to completion. H2O (20 mL) was added to the reaction mixture and the product was subsequently extracted with DCM (10 mL x 3). The organic layers were combined and first washed with a dilute HCl solution (0.2 M, 25 mL), brine (25 mL) and dried over MgSO4 before removing excess solvent under vacuo. The crude product was purified via silica gel flash column chromatography (EtOAc:PET 5:95) to afford compound 4 as a colourless glass (370 mg, 90 %). R = 0.66 (10:90 EtOAc:PET); Mp = 124-128 °C; IR (ATR, cm"1): 3374 (N-H), 2960 and 2873 (C-H), 1727 (C=O), 1529 (arene), 1454 (C=C), 1211 and 1191 (C-O-C), 1005 and 966 (C-N), 757 (C-H); 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 6.86-6.36 (br m, 11H, ArH), 6.13 (br s, 1H, NHR), 4.47 (d, 2H, 2JHH = 13.4 Hz, ArCH^yAr), 4.42 (d, 2H, 2JHH = 13.4 Hz, ArCH^yAr), 3.92-3.76 (m, 8H, OCH2CH2), 3.70(s, 3H, OCH3), 3.16 (d, 2H, 2JHH = 13.4 Hz, ArCH2(a?yAr), 3.13 (d, 2H, 2JHH = 13.4 Hz, ArCH2(a?yAr), 2.00-1.84 (m, 8H, CH2CH2CH3), 1.08-0.94 (m, 12H, CH2CH2CH3); 13C{1H} (75 MHz, CHLOROFORM-rf) Ô ppm 157.0 (ArC), 156.5 (nHCOo), 153.0 (ArC), 135.6 (ArC) 135.5 (ArC), 135.3 (ArC), 135.0 (ArC), 131.5 (ArC), 128.5 (ArC), 128.4 (ArC), 128.1 (ArC), 122.1 (ArC), 121.6 (ArC), 119.4 (ArC), 76.80 (OCH2CH2), 76.76 (0CH2CH2), 52.2 (OCH3), 31.2 (ArCH2Ar), 31.1 (ArCH2Ar), 23.43 (0CH2CH2CH3),23.37(0CH2CH2CH3),23.3 (0CH2CH2CH3),10.6 (0CH2CH2CH3), 10.4 (0CH2CH2CH3); HRMS-Positive: m/z [M+H] + calcd. for C42H52N06: 666.3795; found 666.3782.
(±)-6-Bromo-5-methyl carbamate-25,26,27,28-tetrapropoxy-calix[4]arene (5)
A Schlenk equipped with a magnetic stir bar and flushed with argon was charged with compound 4 (97 mg, 0.15 mmol), NBS (28 mg, 0.16 mmol, 1.1 eq), PTSA (14 mg, 0.071 mmol, 0.5 eq) and Pd(0Ac)2 (1.6 mg, 0.007 mmol, 0.05 eq) in DCE (2 mL). The contents were heated to 60 °C and left to stir for two and a half hours. After the allotted time, the reaction was cooled to room temperature and diluted with DCM (5 mL) before being poured into H20 (10 mL). The product was extracted with DCM (5 mL x 3) and the combined organic layers were subsequently washed with 10 % HCl (10 mL), sat. NaHCO3 (10 mL) and finally brine (10 mL). The solution was then dried over MgS04 and the solvent was removed via reduced pressure. Purification was achieved via silica gel flash column chromatography (2:98 Et0Ac:PET) to yield compound 5 as an amorphous glass (90 mg, 82 %). Rf= 0.70 (DCM); Mp = 174-184 °C; IR (ATR, cm-1): 2957 and 2873 (C-H), 2365 (N-H), 1705 (C=0), 1455 (C=C), 1192 and 1087 (C-O-C), 1005 and 965 (C-N), 762 (C-H); 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 7.86 (br s, 1H, NH), 7.13 (br, 1H, ArH), 7.09 (d, 2H, 2JHH = 7.4 Hz, ArH), 6.90 (t, 1H, 3JHH = 7.4 Hz, ArH) 6.40-6.09 (m, 6H, ArH), 4.49^.36 (m, 4H, ArCH2(alyAr), 4.11-3.81 (m, 4H, 0CH2CH2 and 1H, ArCH2(eq)Ar), 3.83 (s, 3H, 0CH3), 3.74-3.63 (m, 4H, 0CH2CH2), 3.27-3.06 (m, 3H, ArCH2(eq.)Ar), 2.08-1.79 (m, 8H, CH2CH2CH3), 1.10 (t, 3JHH = 7.4 Hz, 6H, CH2CH2CH3), 0.91 (t, 3JHH = 7.4 Hz, 3H, CH2CH2CH3), 0.90 (t, 3JHH = 7.4 Hz, 3H, CH2CH2CH3); 13C{1H} (75 MHz, CHLOROFORM-d) δ ppm 158.0 (ArC), 155.3 (NHCOO), 154.6 (ArC),154.3 (ArC), 137.6 (ArC), 137.1 (ArC), 137.0 (ArC), 136.7 (ArC), 135.5 (ArC), 133.5 (ArC), 132.4 (ArC), 132.1 (ArC), 130.2 (ArC), 129.0 (ArC), 128.9 (ArC), 127.9 (ArC), 127.8 (ArC), 127.5 (ArC), 126.6 (ArC), 122.3 (ArC), 122.3 (ArC), 121.9 (ArC), 121.0 (ArC), 77.1 (0CH2CH2), 77.0 (0CH2CH2), 76.6 (0CH2CH2), 52.6 (0CH3), 31.2 (ArCH2Ar), 31.1 (ArCH2Ar), 31.0 (ArCH2Ar), 30.2 (ArCH2Ar), 23.65 (0CH2CH2CH3), 23.59 (0CH2CH2CH3), 23.1 (0CH2CH2CH3), 23.0 (OCH2CH2CH3), 10.94 (OCH2CH2CH3), 10.91 (OCH2CH2CH3), 9.97 (0CH2CH2CH3), 9.96 (0CH2CH2CH3); HRMS-Positive: m/z [M+NH4]+ calcd. for C42H54BrN206: 761.3165; found 761.3157.
5-Menthyl carbamate-25,26,27,28-tetrapropoxycalix[4]arene (6)
In an oven-dried 2-neck round-bottomed flask, compound 5 (500 mg, 0.823 mmol) was dissolved in DCM (40 mL) and cooled to 0 °C. Pyridine (79.5 //L, 0.987 mmol, 1.2 eq) and menthyl chloroformate (209 //L, 0.987 mmol, 1.2 eq) were subsequently added and the mixture was warmed to room temperature. After 15 min, the contents of the flask were poured into H2O (40 mL) and extracted with DCM (20 mL x 3). The organic layers were then combined and first washed once with dilute HCl solution (25 mL, 0.2 M) followed by brine (25 mL) and finally dried over MgSO4. The solvent was removed under reduced pressure and the crude product was purified via silica gel flash column chromatography (3:97 EtOAc:PET) to afford compound 6 as a colourless glass (650 mg, 98 %). R = 0.47 (10:90 EtOAc:PET) Mp = 68-72 °C, [a]D20.6 = +23.0 ° (c0.017, DCM), IR (ATR, cm"1): 2957, 2923 and 2872 (C-H), 1697 (C = O), 1454 (C=c), 1210 (C-O-C), 1006 and 966 (C-N), 757 (C-H); 1H NMR (600 MHz, CHLOROFORM-rf) Ô ppm 6.84-6.39 (m, 11H, ArH), 6.06 (br. s, 1H, NHR),4.60 (td, 3/HH = 10.8,4.1 Hz, 1H, OCHhexyl) 4.46 (d, 2JHH = 13.4 Hz, 2H, ArCH2(„/Ar), 4.43 (d, 2/HH = 13.4 Hz, 2H, ArCH^yAr), 3.86 (t, 3/HH = 7.6 Hz, 4H, OCH2CH2), 3.82 (t, 3/HH = 7.3 Hz, 2H, OCH2CH2), 3.78 (t, 3/HH = 7.3 Hz, 2H, OCH2CH2), 3.16 (d, 2/HH = 13.4 Hz, 2H, ArCH2(a?yAr), 3.12 (d, 2/HH = 13.6 Hz, 2H, ArCHwAr), 2.10-2.04 (m, 1H, CHC3H7), 1.97-1.85 (m, 8H, CH3CH2CH3), 1.72-1.66 (m, 2H, hexylH), 1.54-1.45 (m, 1H, hexylH), 1.38-1.29 (m, 1H, hexylH), 1.12-0.83 (m, 3H, hexylH), 1.03-0.95 (m, 12H, OCH2CH2CH3), 0.91 (2xd, 3/HH = 6.7 Hz, 6H, CH(CH3)2), 0.79 (d, 2/HH = 7.0 Hz, 3H, CHCH3). 13C{1H} (150MHz, CHLOROFORM-rf) Ô ppm 156.8 (ArC), 156.5 (NHCOO), 152.5 (ArC), 135.4 (ArC), 135.2 (ArC), 134.9 (ArC), 131.8 (ArC), 128.31 (ArC), 128.27 (ArC), 127.9 (ArC), 122.92 (ArC), 122.91 (ArC), 121.5 (ArC), 118.5 (ArC) 76.66 (OCH2CH2), 76.64 (OCH2CH2), 76.62 (OCH2CH2), 74.5 (OCHhexyl), 47.4 (hexylC), 41.4 (hexylC), 34.3 (hexylC), 31.4 (ArCH2Ar), 31.1 (ArCH2Ar), 31.0 (ArCH2Ar), 26.2 (CH(CH3)2), 23.5 (hexylC), 23.3 (OCH2CH2CH3), 23.21 (OCH2CH2CH3), 23.19 (OCH2CH2CH3), 22.1 (CH(CH3)2), 20.9 (CH(CH3)2), 16.4 (hexylCH3), 10.43 (OCH2CH2CH3), 10.41 (OCH2CH2CH3), 10.2 (OCH2CH2CH3). HRMS-Positive: m/z [M+NH4]+ calcd. for C51H71N2O6: 807.5312; found 807.5313.
4-Bromo-5-menthyl carbamate-25,26,27,28-tetrapropoxycalix [4]arene (7a and 7b)
An oven-dried 5 mL Schlenk equipped with a magnetic stir bar was charged with NBS (14.9 mg, 0.084 mmol, 1.1 eq), PTSA-H2O (7.2 mg, 0.038 mmol, 0.5 eq), Pd(OAc)2 (0.9 mg, 0.004 mmol, 0.05 eq) and DCM (1 mL). The reaction vessel was subjected to five freeze pump thaw cycles before being flushed with argon and cooled to -35 °C. Once cooled, compound 10 (60 mg, 0.078 mmol) was added at once and the reaction was left to stir at -35 °C for a further 4 h. After the time had elapsed, the reaction contents were diluted with DCM (10 mL) and washed with a 10 % HCl solution (15 mL). The aqueous phase was extracted with DCM (10 mL x 3) before combining the organic phase and washing it once with sat. NaHCO3 (20 mL) and finally brine (20 mL). The solution was finally dried over MgSO4 and concentrated under vacuo. The crude product was then purified via silica gel flash column chromatography (1:99 EtOAc:PET) to produce compounds 7a and 7b as an inseparable mixture of diastereomers (61 mg, 91 %). R = 0.27 (4:96 EtOAc:PET); Mp = 74-84 °C; IR (ATR, cm-1): 3411 (N-H), 2957,2931 and 2872 (C-H), 1732 (C=O), 1509 (arene), 1455 (C=C), 1209 and 1187 (C-O-C), 1005 and 966 (C-N), 756 (C-H); 1H NMR (300 MHz, CHLOROFORM-d) δ ppm 7.95-7.84 (m, 1H, ArH - meta to Br), 7.20-7.02 (m, 2H, ArH), 6.91 + 6.89 (2 x t, 3JHH= 7.3 Hz, 1H, ArH), 6.36-6.06 (m, 6H, ArH), 4.77-4.61 (m, 1H, menthyl-OCH), 4.48^.36 (m, 4H, ArCH^yAr), 4.10-3.79 (m, 4H, OCH2CH2 + 1H, ArCH2(eq)Ar), 3.72-3.61 (m, 4H, OCH2CH2), 3.24-3.09 (m, 3H, ArCH2(a?yAr), 2.23-2.03 (m, 1H, menthylH), 2.01-1.80 (m, 8H, OCH3CH2CH3), 1.72 (app d, 2JHH= 11.6 Hz, 2H, menthyl-H), 1.51-1.38 (m, 1H, menthyl-H), 1.51-0.74 (m, 27H, menthyl-H's, menthyl-CH3's, OCH3CH2CH3); 13C{1H} (75 MHz, CHLOROFORM-d)* δ ppm 158.1 + 157.1 + 155.3 (ArC-OPr), 154.4 (ArC-OPr Br ring), 153.6 (C=O), 139.2 + 139.0 + 137.5 + 137.2 + 136.7 + 136.4 + 133.5 + 132.7 + 132.5 + 132.4 + 132.2 (ArC), 131.3 (ArCH), 130.7 + 130.6 (ArC-Br), 129.0 + 127.9 + 127.7 + 127.5 + 127.3 + 126.7 + 126.5 + 122.3 + 121.9 (ArCH), 120.8 (ArCH - meta to Br), 114.4 + 113.9 (ArC), 77.0 + 76.6 (OCH2CH2CH3), 75.5 + 75.4 (menthyl-OCH), 47.3 (menthyl-CH), 41.4 (menthyl-CH2), 34.4 (menthyl-CH2), 31.6 + 31.0 + 30.3 (ArCH2Ar), 26.4 (menthyl-CH), 23.6 + 23.0 (menthyl-CH2 + OCH2CH2CH3), 22.2 + 21.0 + 16.7 (menthyl-CH3's), 10.9 + 9.9 (-OCH2CH2CH3); HRMS-Positive: m/z [M+NH4]+ calcd. for C51H70BrN2O6: 885.4417; found 885.4427.
4-Bromo-5-amino-25,26,27,28-tetrapropoxycalix[4]arene (8)
In an oven-dried 2-neck round-bottomed flask equipped with a reflux condenser and magnetic stir bar was charged with calixarene 7a and 7b (110 mg, 0.127 mmol), a 1 M solution of TBAF in THF, (1.27 mL, 10 eq) and THF (10 mL). The contents were heated to reflux and left to stir for 36 h. After the time had elapsed, the solution was concentrated and then diluted with EtOAc (15 mL) before being washed with H2O (10 mL x 6) and finally brine (10 mL). The liquid was dried over MgSO4 and concentrated via reduced pressure. The crude product was then purified firstly via silica gel flash column chromatography (EtOAc:PET 1:9) and then heated at 80 °C under reduced pressure for 8 h to afford compound 8 as a yellow solid (72 mg, 83 %).
Rf= 0.33 (5:95 EtOAc:PET); Mp = 130-136 °C; IR (ATR, cm-1): 1264 (C-N), 731 (C-H), 703 (C-H); 1H NMR (600 MHz, CHLOROFORM-d) δppm 7.09 (d, 3JHH = 7.4 Hz, 2H, ArH), 6.91 (t, 3JHH = 7.4 Hz, 1H, ArH), 6.60 (s, 1H, ArH), 6.30-6.12 (m, 6H, ArH), 4.45 (d, 2JHH = 13.3 Hz, 1H, ArCH^yAr), 4.44 (d, 2JHH = 13.3 Hz, 1H, ArCH^yAr), 4.37 (d, 2JHH= 13.6 Hz, 2H, ArCH^yAr), 4.06-3.79
(m, 4H, OCH2CH2,2H,NH2), 3.83 (d, 2JHH = 13.6 Hz, 1H, ArCH2(eq)Ar), 3.75-3.62 (m, 4H, OCH2CH2), 3.16 (d, 2JHH= 13.4 Hz, 1H, ArCH2(eq)Ar), 3.15 (d, 2JHH= 13.4 Hz, 1H, ArCH2(eq)Ar), 3.03 (d, 2JHH = 13.3 Hz, 1H, ArCH2(eq)Ar), 2.04-1.82 (m, 8H, CH3CH2CH3), 1.10 (t, 3JHH = 7.4 Hz, 6H, CH2CH2CH3), 0.90 (t, 3JHH = 7.4 Hz, 3H, CH2CH2CH3), 0.89 (t, 3JHH = 7.4 Hz, 3H, CH2CH2CH3); 13C{1H} (150 MHz, CHLOROFORM-d) δ ppm 158.1 (ArC), 155.3 (ArC), 150.9 (ArC), 139.0 (ArC), 137.6 (ArC), 137.3 (ArC), 137.1 (ArC), 136.6 (ArC), 133.5 (ArC), 133.3 (ArC), 132.6 (ArC), 129.0 (ArC), 128.9 (ArC), 127.7 (ArC), 127.6 (ArC), 127.3 (ArC), 126.8 (ArC), 122.2 (ArC), 121.8 (ArC), 115.7 (ArC), 110.4 (ArC), 77.06 (OCH2CH2), 76.96 (OCH2CH2), 76.95 (OCH2CH2), 76.55 (OCH2CH2), 31.18 (ArCH2Ar), 31.11 (ArCH2Ar), 30.89 (ArCH2Ar), 29.93 (ArCH2Ar), 23.66 (OCH2CH2CH3), 23.62 (OCH2CH2CH3), 23.10 (OCH2CH2CH3), 22.97 (OCH2CH2CH3), 10.95 (OCH2CH2CH3), 10.94 (OCH2CH2CH3), 10.01 (OCH2CH2CH3), 9.99 (OCH2CH2CH3). HRMS-Positive: m/z [M+H]+ calcd. for C40H49BrNO4: 686.2845; found 686.2849.
Acknowledgements
This work was supported by the National Research Foundation (Grant CPRR160428163281) and Stellenbosch University. K.J.V is grateful to the DST/NRF for an MSc innovation scholarship and L.H. to the NRF for postdoctoral funding. We would also like to thank Dr. J. Brand and Ms. E Malherbe for the NMR spectroscopic service and Dr. M Stander and Mr. M Taylor for the mass spectroscopic service.
* Dueto diastereomers, signals were difficult to assign accurately. Assignments based on 2D NMR spectroscopy only where it was clear.
ORCID iDs
KJ.Visagie: orcid.org/0000-0002-3079-5049
L.Hodson: orcid.org/0000-0001-9779-1133
G.E.Arnott: orcid.org/0000-0002-6872-123X
References
1 A. Szumna, Inherently chiral concave molecules - from synthesis to applications, Chem. Soc. Rev., 2010, 39(11), 4274^285. [ Links ]
2 G.E.Arnott, Inherently chiral calixarenes: synthesis and applications, Chem. -A Eur. J., 2018, 24(8), 1744-1754. [ Links ]
3 L.X. Dai, T. Tu, S.L. You, W.P. Deng and X.L. Hou, Asymmetric catalysis with chiral ferrocene ligands, Acc. Chem. Res., 2003,36(9), 659-667. [ Links ]
4 S.A. Herbert and G.E. Arnott, An asymmetric ortholithiation approach to inherently chiral calix[4]arenes, Org. Lett., 2009,11(21), 4986-989. [ Links ]
5 S.A. Herbert and G.E. Arnott, Synthesis of inherently chiral calix[4]arenes: stereocontrol through ligand choice, Org. Lett., 2010, 12(20), 4600-4603. [ Links ]
6 S.A. Herbert, D.C. Castell, D.C. Clayden and G.E. Arnott, Manipulating the diastereoselectivity of ortholithiation in planar chiral ferrocenes, Org. Lett., 2013,15(13), 3334-3337. [ Links ]
7 D.C. Castell, N. Lesotho, VI. Nikolayenko and G.E. Arnott, Inherently chiral calix[4]arenes: a chiral sulfoxide as an ortholithiation director, European J. Org. Chem., 2017, 2017(29), 4328^333. [ Links ]
8 F.M. Moghaddam, G. Tavakoli, B. Saeednia, P. Langer and B. Jafari, Palladium-catalyzed carbamate-directed regioselective halogena-tion: a route to halogenated anilines, J. Org. Chem., 2016, 81(9), 3868-3876. [ Links ]
9 G.E. Arnott, Inherently chiral calixarenes: synthesis and applications, Chem. -A Eur. J., 2018, 24(8), 1744-1754. [ Links ]
10 M.C. Davis, Chlorination of aniline and methyl carbanilate by N-chlorosuccinimide and synthesis of 1,3,5-trichlorobenzene, Synth. Commun, 2009, 39(6), 1100-1108. [ Links ]
11 P.V.N. Reddy, B. Banerjee andM. Cushman, Efficient total synthesis of ammosamide B, Org. Lett., 2010,12(13), 3112-3114. [ Links ]
12 A. Roth, H. Li, C. Anorma and J. Chan, A reaction-based fluorescent probe for imaging of formaldehyde in living cells, J. Am. Chem. Soc., 2015,137(34), 10890-10893. [ Links ]
13 D.L. Boger, R.J. Wysocki, S.A. Munk, T. Ishizaki, P.A. Kitos and O. Suntornwat, Total synthesis and evaluation of (±)-N-(tert-butyloxy-carbonyl)-CBI, (±)-CBI-CDPI1, and (±)-CBI-CDPI2: CC-1065 functional agents incorporating the equivalent 1,2,9,9a-tetrahydrocyclo-prop[1,2-c]benz[1,2-e]indol-4-one (CBI) left-hand subunit, J. Am. Chem. Soc., 1989,111(16), 6461-6463. [ Links ]
14 M. Guyonnet and O. Baudoin, Synthesis of tricyclic nitrogen heterocycles by a sequence of palladium-catalyzed N-H and C(sp 3)-H arylations, Org. Lett., 2012,14(1), 398-01. [ Links ]
15 R.A. Fujimoto, L.W. Mcquire, L.G. Monovich, B.B. Mugrage, D.T. Parker, J.H. Van Duzer and S. Wattanasin, Substituted amino phenylacetic acids, derivatives thereof, their preparation and their use as cyclooxygenase 2 (cox-2) inhibitors, WO 2004/048314 A1,2004. [ Links ]
16 N. Uhlig and C.J. Li, Aniline carbamates: a versatile and removable motif for palladium-catalyzed directed c-h activation, Chem. - A Eur. J., 2014, 20(38), 12066-12070. [ Links ]
17 X. Shi and D. Shi, Recent advances in transition-metal-catalyzed halides formation, Curr. Org. Chem., 2018, 22(23), 2229-2255. [ Links ]
18 C. Que, N. Chen and J. Xu, Application of carbamates in the C-H bond activation, Prog. Chem., 2018, 30(2-3), 139-155. [ Links ]
19 R. Das and M. Kapur, Transition-metal-catalyzed site-selective C-H halogenation reactions, Asian J. Org. Chem., 2018, 7(8), 1524-1541. [ Links ]
20 C. Sambiagio, D. Schõnbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M.F. Zia, J. Wencel-Delord, T. Besset, B.U.W. Maes and M. Schnürch, A comprehensive overview of directing groups applied in metal-catalysed C-H functionalisation chemistry, Chem. Soc. Rev., 2018, 47(17), 6603-6743. [ Links ]
21 A.M.A. Van Wageningen, E. Snip, W. Verboom, D.N. Reinhoudt and H. Boerrigter, Synthesis and application of iso(thio)cyanate-functionalized calix[4]arenes, Liebigs Ann., 1997,11, 2235-2245. [ Links ]
22 C.D. Jurisch and G.E. Arnott, Attempted synthesis of a meta -meta-lated calix[4]arene, Beilstein J. Org. Chem., 2019,15(1), 1996-2002. [ Links ]
23 S. Pinchas and D. Ben-Ishai, The carbonyl absorption of carbamates and 2-oxazolidones in the infrared region, J. Am. Chem. Soc., 1957, 79(15), 4099-4104. [ Links ]
24 U. Jacquemard, V. Bénéteau, M. Lefoix, S. Routier, J.Y. Mérour and G. Coudert, Mild and selective deprotection of carbamates with Bu 4 NF, Tetrahedron, 2004, 60(44), 10039-10047. [ Links ]
25 P.M. Esch, H. Hiemstra and W.N. Speckamp, Oxazinone versus allene formation in the reaction of N-alkoxycarbonyliminium ions with propargyltrimethylsilane, Tetrahedron Lett., 1988, 29(3), 367-370. [ Links ]
Received 12 September 2019
Revised 14 November 2019
Accepted 10 January 2020
* To whom correspondence should be addressed. E-mail: arnott@sun.ac.za
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]


{kind=link}