










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.72 Durban 2019
http://dx.doi.org/10.17159/0379-4350/2019/v72a23
RESEARCH ARTICLE
Molecular Structure, FT-IR, NMR (13C/1H), UV-Vis Spectroscopy and DFT Calculations on (2Z, 5Z)-3-N(4-Methoxy phenyl)-2-N'(4-methoxy phenyl imino)-5-((E)-3-(2-nitrophenyl)allylidene) thiazolidin-4-one
Rachida RahmaniI, II; Ahmed DjafriI, III; Abdelkader ChouaihI*; Ayada DjafriIV; Fodil HamzaouiI, V; Abdelghani M. KrallafaVI
ILaboratory of Technology and Solid Properties (LTPS), Abdelhamid Ibn Badis University - Mostaganem, 27000 Mostaganem, Algeria
IIDepartement de Génie des Procedes, Centre Universitaire Ahmed Zabana - Relizane, 48000 Relizane, Algeria
IIICentre de Recherche Scientifique et Technique en Analyses Physico-chimiques (CRAPC), BP 384-Bou-Ismail-RP 42004, Tipaza-Algeria
IVLaboratoire de Synthèse Organique Appliquée (LSOA), Departement de Chimie, Faculte des Sciences, Université d'Oran 1 - Ahmed Ben Bella, 31000 Oran, Algeria
VLPFM Académie de Montpellier, France
VILCPM, Department of Chemistry, Faculty of Sciences, University of Oran 1, Ahmed Benbella, 31000, Algeria
ABSTRACT
In this study, some molecular properties of (2Z, 5Z)-3-N(4-methoxy phenyl)-2-N'(4-methoxy phenyl imino)-5-((E)-3-(2-nitro-phenyl) allylidene) thiazolidin-4-one (MNTZ) are evaluated using a combination of spectroscopic characterization (FT-IR, 1H and 13C NMR chemical shifts) and theoretical calculations. Molecular geometry, vibrational wavenumbers, gauge-independent atomic orbital (GIAO), 1H and 13C chemical shift values and NBO analysis are investigated using B3LYP and PBE functionals with the 6-31G(d,p) basis set in the ground state. The calculated geometrical parameters and vibrational spectra are compared to available experimental data and each vibrational frequency is assigned on the basis of potential energy distribution (PED). The electronic transitions are calculated using time-dependent density functional theory (TDDFT). The energy band gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies are obtained by computing the frontier molecular orbitals using the B3LYP/6-31G(d,p) and PBE/6-31G(d,p) levels along with the global reactivity descriptors. Mulliken atomic charges and molecular electrostatic potential (MEP) are simulated using both functionals to find more reactive sites for electrophilic and nucleophilic attack. Finally, the thermodynamic functions (heat capacity, entropy, and enthalpy) from spectroscopic data are obtained and discussed in the range of 100-1000 K.
Keywords: FT-IR, UV-Vis, HOMO-LUMO, thiazolidinones, DFT, NMR spectra, NBO.
1. Introduction
It is well known that heterocyclic compounds are one of the most important classes of organic compounds. During the last few decades, they have attracted attention due to their proven usefulness in material sciences.1-3 Moreover, the study of heterocyclic compounds is also of great interest theoretically. Specifically, thiazolidinones which belong to an important group of heterocyclic compounds, have been widely explored for their application in the field of medicine.4,5 Thiazolidinones derivatives, particularly 4-thiazolidinones gained the attraction of researchers as a result of their broad-spectrum biological activity.6-8 In addition, thiazolidinones are also known to exhibit push-pull effects and have photovoltaic applications due to the substituent at position 5 of the thiazole moiety.9,10 On the other hand, the presence of heteroatoms nitrogen, oxygen or sulfur incorporated into the heterocyclic rings may have a strong effect on the polarity and polarizability of the molecule, the angle of rotation between its fragments, the planarity and thus on its stability.
In this paper, our interest was the thiazolidinones derivative, containing delocalized electrons. The π-conjugated system allows the title compound to exhibit the asymmetric electronic distribution which leads to increased charge transfer. Recently, the synthesis and X-ray crystal structure of the title compound was published.11
Based on these studies, and as a continuation of our previous work on thiazolidinone compounds12-15, herein, we report the optimized molecular structure as well as a detailed spectroscopic study on (2Z, 5Z)-3-N(4-methoxy phenyl)-2-N'(4-methoxy phenyl imino)-5-((E)-3-(2-nitrophenyl) allylidene) thiazolidin-4-one (MNTZ) using IR, 1H and 13C NMR spectra. The natural bond orbital (NBO) analysis is carried out to interpret intramolecular charge transfer (ICT). Besides considering charge transfer within this molecule, we have determined HOMO-LUMO orbitals and global reactivity descriptors. The reactivity and stability of MNTZ were determined by global chemical parameters such as ionization potential, the electron affinity, the absolute electronegativity, electrophilicity index, the absolute hardness and softness. The ionization potential represents the amount of energy required to remove an electron from an isolated atom or molecule where the electron affinity refers to the capability to accept one electron from a donor. These two parameters can be obtained from HOMO-LUMO analysis. The power of an atom in molecule to attract electrons to itself describes its electronegativity and the electrophilicity is defined as a measure of the propensity of a species to accept electrons. The electrophilicity index was calculated using the electronic chemical potential and chemical hardness. A good, more reactive, nucleophile is characterized by a lower value of chemical potential and electrophilicity index, and conversely a good electro-phile is characterized by a high value of chemical potential and electrophilicity index. The UV-Visible spectroscopic studies along with the calculated value of the band-gap energy were used to interpret charge transfer within the molecule. Mulliken population analysis, molecular electrostatic potential (MEP) and thermodynamic parameters were investigated using B3LYP and PBE functionals in the gas phase.
2. Experimental and Computational Details
Synthesis, solid state structure and spectral data (IR, 1H NMR, 13C NMR) of the title compound are reported in our previous work.11 IR spectra were recorded in KBr pellet on a JASCO FT/IR 4210 Fourier Transform Infrared Spectrometer and the reported wave numbers were given in cm-1. The 1H NMR and 13C NMR were recorded on Brüker Ac DPX-200(300 MHz) spectrometer in CDCl3 as solvent using tetramethylsilane as an internal reference standard.
Density functional theory (DFT) has proved to be extremely useful for studying the electronic structures of molecules. Calculations of the new (2Z, 5Z)-3-N'(4-methoxy phenyl)-2-N'(4-methoxy phenyl imino)-5-((E)-3-(2-nitrophenyl) allylidene) thiazolidin-4-one (MNTZ) are carried out using the Gaussian 09 program and GaussView molecular visualization software.16,17 The parent structure for the calculations is obtained from the X-ray coordinates11 and this structure is optimized at the DFT level of theory with the B3LYP18,19 and PBE20 functionals and the 6-31g(d, p) basis set. Optimized structures are used in the vibra-tional frequency calculations to ensure that the obtained structures represent a local minima. The theoretical vibrational spectrum is interpreted through the Potential Energy Distribution (PED) using Vibrational Energy Distribution Analysis (VEDA) program.21 The nuclear magnetic resonance (NMR) chemical shift calculations are undertaken using the Gauge-Independent Atomic Orbital (GIAO) method at B3LYP/6-31G(d,p) level.
Chloroform solvent effects on theoretical NMR parameters were included using single point calculation on optimized gas phase geometry.
The time-dependent density functional theory (TD-DFT) method on the ground state was used to calculate the excited states and the electronic transitions. The chloroform solvent effect has been considered using the Polarized Continuum Model (PCM).22 The vertical transition wavelength/energies are calculated for 10 excited singlet states by the TD-DFT methodology using the same functionals as geometry optimization. The NBO analysis is performed with DFT/B3LYP/6-31g(d,p) level to elucidate the conjugation, charge transfer and delocalization of electron density within the molecule. Besides, Mulliken and NBO atomic charges, the molecular electrostatic potential (MEP), frontier molecular orbital (FMOs), the dipole moment and thermodynamic properties of MNTZ were investigated using both functionals.
3. Results and Discussion
3.1. Optimized Geometry
The optimized structure of MNTZ molecule is given in Fig. 1 with atomic labelling and the corresponding cartesian/internal coordinates are reported in Table S1 (see supplementary information). The geometrical parameters (bond lengths, bond angles, and dihedral angles) computed by B3LYP and PBE functionals with the 6-31G(d,p) basis sets are listed in Tables S2, S3 and S4 (see supplementary information), together with the X-ray parameters.11 The differences between calculated and experimental bond lengths and angles are within a few Angstroms and degrees, respectively, when compared to the experimental parameters, which indicate that our calculations are acceptable. The small differences can be due to the fact that calculated data are collected in the gas phase, while the experimental data are acquired in the solid state.
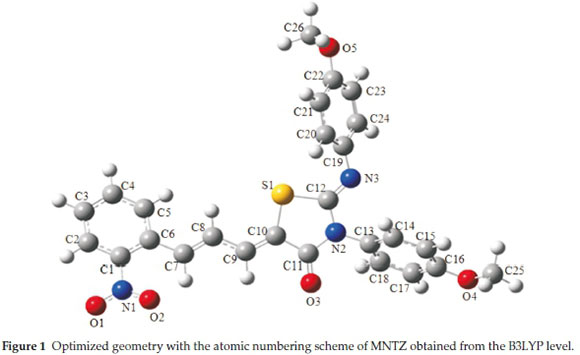
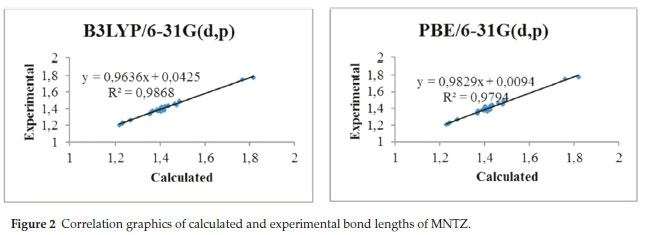
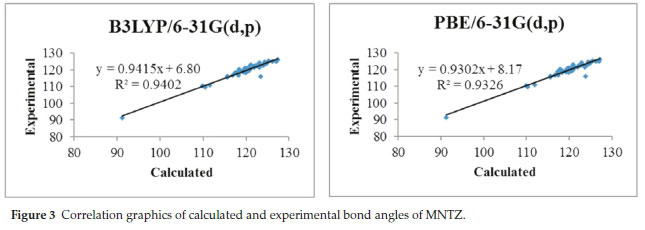
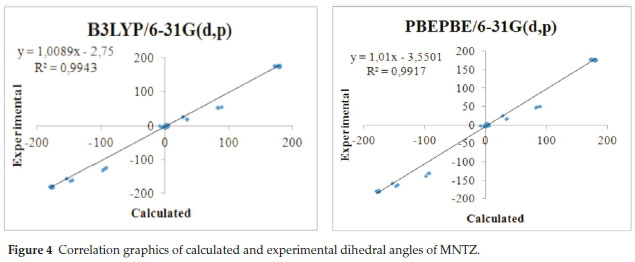
To get a better comparison of the geometrical parameters, correlation graphs between the calculated and the experimental parameters of bond lengths, bond angles and dihedral angles are shown in Fig. 2, Fig. 3 and Fig. 4, respectively. The correlation values R2 obtained by B3LYP and PBE functionals with 6-31g(d,p) basis set are 0.9868 and 0.9794 for bond lengths, 0.9402 and 0.9326 for bond angles and 0.9943 and 0.9917 for dihedral angles. These results confirmed the good agreement between calculated and experimental parameters.
3.2. Vibrational Assignments
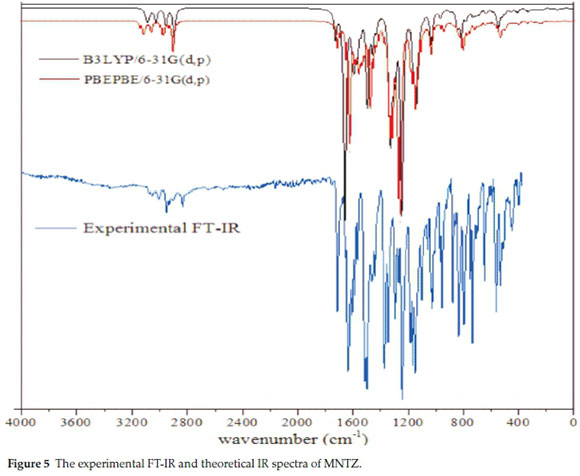
The experimental FT-IR and the calculated wavenumbers by B3LYP and PBE methods with 6-31g(d,p) basis set and their assignment using potential energy distribution (PED) calculation are given in Table S5 (see supplementary information). The calculated and experimental infrared spectra are shown in Fig. 5. No imaginary frequency has been found, which means that the optimized geometry is located at the local lowest point on the potential energy surface. The calculated frequencies are higher than the experimental values for the majority of the normal modes, for this reason, the scaling factors 0.961 and 0.986 are used for B3LYP and PBE calculated frequencies23,24, respectively. The MNTZ consists of 56 atoms and hence it shows 162 (3N-6) normal modes of vibration25 active in infrared absorption.
3.2.1. C-H Vibrations
The C-H stretching vibrations of aromatic compounds appear in the range of 3100-3000 cm-1.26 In the present study, the phenyl ring CH stretching vibrations are predicted at the region 3120-3070 cm-1 for B3LYP and 3128-3084 cm-1 for PBE, which are in good agreement with the observed value of 3057.7 and 3008.8 cm-1 in FT-IR spectrum.
The experimental wavenumbers of CH stretching vibrations of allylidene fragment are observed at 2951 and 2930 cm-1 in the FT-IR spectrum. The corresponding theoretical values are 30583039 cm-1for B3LYP and 3064-3048 cm-1 for PBEPBE functional.
The in-plane C-H bending vibration occurs in the region 1300-1000 cm-1.27 This vibration is computed in the range 1460-985 cm-1 for B3LYP and 1448-978 cm-1 for PBE and the corresponding experimental values are found in the region 1296-1009 cm-1 in FT-IR. The out-of-plane C-H bending vibrations give rise to bands in the region 900-625 cm-1.28 This vibration is predicted at 967-520 and 950-512 cm-1 with B3LYP and PBE levels, respectively, and observed at 968-647 cm-1 in FT-IR.
3.2.2. C=C Vibrations
Generally, the C=C stretching vibrations in aromatic compounds occur in the region 1650-1430 cm-1.29 In the present study, the C=C stretching vibration of benzene rings appears at 1637, 1608, 1513.7 and 1439.8 cm-1 in the FT-IR spectrum. The corresponding computed values are 1609,1542 and 1423 cm-1 for ring 1 (C1-C6), 1605,1573 and 1409 cm-1 for ring 2 (C13-C18) and 1601,1557and 1404 cm-1 for ring3 (C19-C24). On the other hand, the band observed at 1587 cm-1 in the FT-IR spectrum is assigned to C=C stretching vibration of allylidene fragment. The corresponding predicted values with the B3LYP level are 1593 and 1583 cm-1 for C7=C8 and C9=C10, respectively.
3.2.3. C=N Vibration
The observed frequency at 1654 cm-1 in the infrared spectrum is assigned to C=N stretching vibrations. This vibration appears at 1658 cm-1 in theoretical IR computed with B3LYP/6-31g(d,p) level and shows a pure mode and its PED contribution is about 74 %.
3.2.4. Nitro Group Vibrations
The asymmetrical stretching vibrations of substituted nitrobenzene occur in the region 1560-1490 cm-1,30 and symmetric stretching vibrations occur in the region 1370-1310 cm-1. The FT-IR spectrum for MNTZ gives peaks at 1566.1 and 1374.4 cm-1, and these peaks are assigned as asymmetric and symmetric NO2 stretching vibrations. These peaks are predicted at 1566 and 1339 cm-1 for B3LYP and 1551, 1353 cm-1 for PBE, respectively.
The asymmetric NO2 stretching vibration calculated by B3LYP is found to be exactly correlated with the experimental one. The observed NO2 in-plane bending vibration is assigned at 841cm-1 in the FT-IR spectrum. The wavenumbers are computed at 842 and 814 cm-1 for the scissoring modes which are correlated with the experimental values. Furthermore, the out-of-plane bending vibration is observed at 683 in the FT-IR spectrum. The corresponding calculated values are 771 and 682 cm-1 with the B3LYP level.
3.2.5. Methoxy Group Vibrations
The asymmetric and symmetric CH stretching vibrations of the two methyl groups occur in the region 3029-2954 and 2900-2896 cm-1 for B3LYP/6-31g(d,p) level, respectively. These vibrations are observed at 2930-2834 cm-1 in the FT-IR spectrum. The in-plane bending modes of CH3 appear at 1461 cm-1 in the FT-IR spectrum. The calculated values at 1460 and 1430 cm-1 obtained by B3LYP level are consistent with FT-IR results. Moreover, the out-of-plane bending vibrations are measured at 1166 in FT-IR and computed at 1165 and 1133 cm-1 with B3LYP/6-31g(d,p). The bands calculated in the range 1036-1035, 280-227 and 156-85 cm-1 are assigned as the stretching O-C, in-plane and out of plane C-OCH3 vibrations.
3.2.6. Thiazolidinone Group Vibrations
Vibrational analysis of thiazolidinone group is made on the basis of C=O, C-N, and C-S vibrations. The carbonyl stretching vibration is observed at 1713 cm-1 in the experimental infrared spectrum and calculated at 1725 and 1711 cm-1 with B3LYP and PBE levels, respectively. The stretching C-N vibration gives rise to a band at 1345 cm-1 in the FT-IR spectrum and the corresponding calculated values are 1327 and 1351 cm-1 with B3LYP and PBEPBE levels, respectively. The bands observed at 591 and 446.9 cm-1 in the IR spectrum are assigned to stretching vC-S and bending áCSC, respectively. These bands are calculated at 588 and 446 cm-1 with the B3LYP level. All these results are in good agreement with the literature.31-33
3.3. 1H and 13C NMR Spectral Analysis
The theoretical 1H and 13C NMR isotropic shielding were com-
puted with the B3LYP/ 6-31G(d,p) level using chloroform as a solvent and the gauge-independent atomic orbital (GIAO) method.34 The TMS shieldings with B3LYP/GIAO/6-31G(d,p) are 31.74 ppm and 192.12 ppm for 1H and 13C NMR, respectively.35 The chemical shift of the studied molecule is δ(MNTZ) where = σ(IMS)=σ(MNTZ)(σ is a chemical shielding).361H and 13C NMR spectra of MNTZ are shown in Fig. 6 and Fig. 7, srespectively. The experimentaland calculated1H and 13C isotropic chemical shifts for MNTZ using the B3LYP/6-31G(d,p) level of theory are given in Table S6 (see supplementary information).
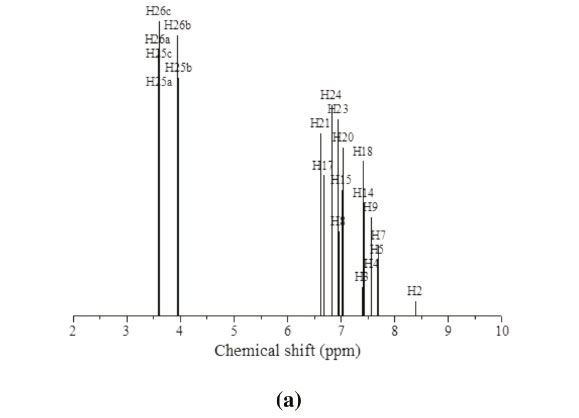
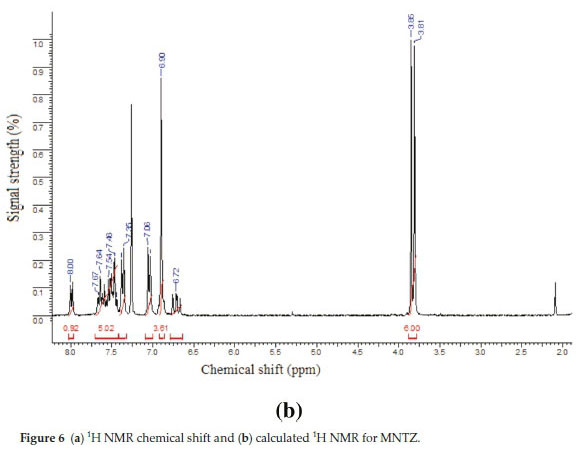
The 1HNMRspectrum (Fig. 6a) of MNTZ in chloroform shows two singlet peaks at 3.81 and 3.85 ppm for the methoxy moieties, the corresponding calculated ranges (Fig. 6b) are 3.58-3.95 ppm for O25CH3 and 3.61-3.97 ppm for O26CH3. The aromatic ring protons give signals in the range of 6.9-8 ppm. The singlet peak at 8 ppm is assigned to the H2 proton (calculated value as 8.4 ppm). Also, the singlet peakat 6.9 ppm is assigned to phenyl protons (H15,H17,H21,andH23),the correspondingcalculated values are 7.0, 6.8, 6.9 and 6.8 ppm, respectively. The doublet peak at 7.04 ppm is assigned to H20 and H24 protons and the doublet peak at 7.35 ppm is assigned to H14 and H18 protons which correlated with the calculated value of 7.4 ppm. The multiplet between 7.43-7.67 ppm is assigned to H3, H4 and H5 protons. For the allylidene fragment protons, H8 appears as a doublet of doublets at 6.71 ppm (calculated at 6.95 ppm) while H7 and H9 appear as multiplet in 7.43-7.67 ppm range, corresponding calculated values at 7.72 and 7.55 ppm, respectively.
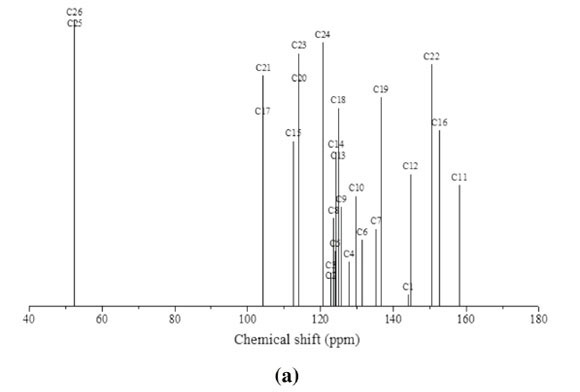
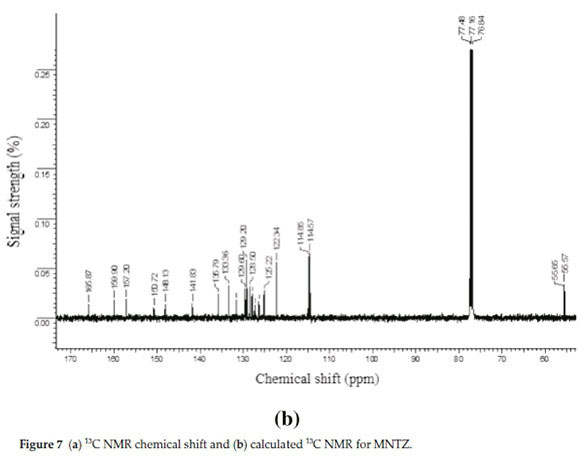
The experimental and calculated 13C NMR spectra of MNTZ in chloroform are found at the range 55-165 ppm and 52-159 ppm and are shown in Fig. 7a and Fig. 7b, respectively. Among the carbon atoms, C11 gives the highest NMR chemical shift which is about 165.87 ppm, the corresponding estimated value is 159.32 ppm due to the effect of carbonyl moiety. The chemical shifts for C16 (observed at 159.9 and estimated at 153.4 ppm) and C22 (observed at 157.2 and estimated at 151.1 ppm) are found to be higher than the other phenyl carbons due to the electro-negativity of the oxygen atoms. Also, the experimental signals of C1 and C12 appear in the higher frequency region at 148.13 and 150.72 ppm, respectively, the corresponding calculated values are 144.74 and 146.01 ppm, respectively, due to the nitrogen atoms. Theotherphenylcarbons NMRpeaks areobservedinthe region of 114-135 ppm whereas the calculated ones range between 105 and 136 ppm. The signals for the allylidene fragment carbons (C7, C8, and C9) are observed at 141.8, 125.2 and 129.6 ppm while the calculated values are 136.9, 123.6 and 127.0 ppm, respectively. The carbon atoms C25 and C26 of the methoxy group give peaks at 55.65 and 55.57 ppm which is in agreement with the calculated values of 53.01 and 52.85 ppm, respectively. All these results show that the B3LYP functional with the GIAO method and 6-31G(d,p) basis set, using chloroform as solvent predicted well the 1H and 13C NMR spectra.
3.4. Natural Bond Orbital (NBO) Analysis
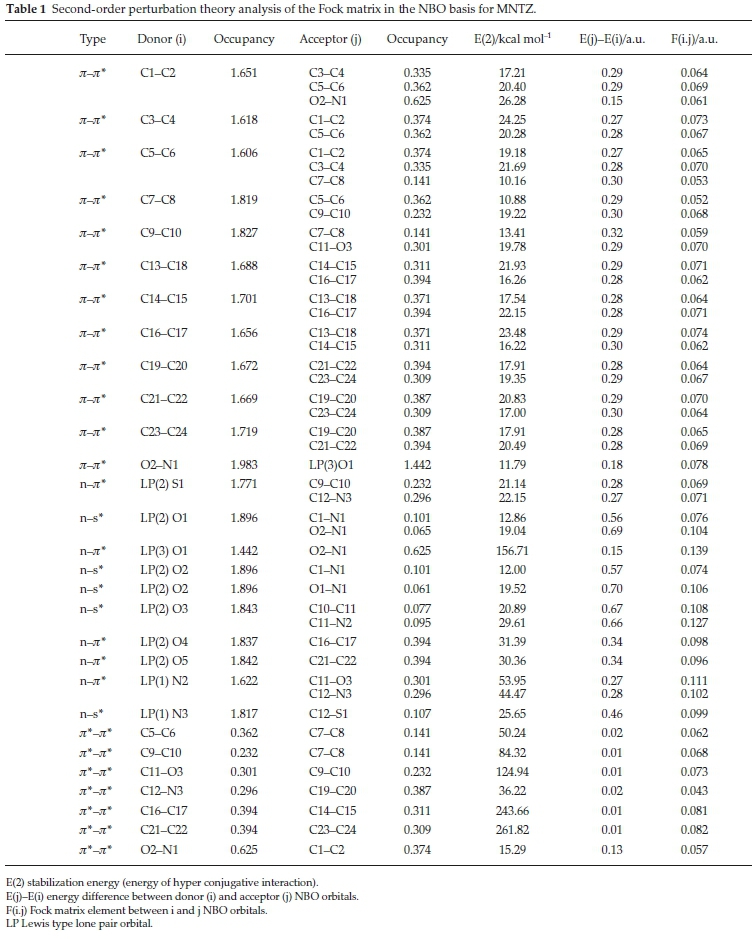
NBO analysis has been performed using NBO 3.137 implemented in the Gaussian 09 program at DFT/B3LYP/6-31G(d,p) level to elucidate the conjugation, charge transfer and delocalization of electron density within the molecule.38 In NBO analysis large E(2) value shows intensive interaction between electron-donors and electron acceptors and therefore indicating a greater extent of conjugation in the system.39 The intramolecular hyper-conjugative interactions are formed by the orbital overlap between bonding jr(C-C) and antibonding jr*(C-C) orbitals. These interactions have greater energy contributions from 10.16 to 24.25 Kcal mol-1 as depicted in Table 1. The intramolecular interactions are also due to the overlap between lone pair of oxygen, nitrogen and sulfur atoms and antibonding of (C-C), (C-N), (O-N) and (C-O) orbitals. For example, the strong intramolecular hyperconjugative interaction n(O1) - 7t*(O2-N1) leads to stabilization energy of 156.71 Kcal mol-1. Furthermore, the electron lone pair in the nitrogen of thiazoli-dinones ring LP(1) N2 transfer its electron to antibonding orbital jr*(C11-O3) and jr*(C12-N3) with stabilization energy of 53.95 and 44.47 Kcal mol-1, respectively. Besides, the most important interactions are mainly from jr*(C11-O3), jr*(C21-C22) and jr*(C16-C17) orbitals as donor to the antibonding jr*(C9-C10), jr*(C23-C24) and jr*(C14-C15) orbitals as acceptor with a strong stabilization energy of 124.94, 261.82 and 243.66 Kcal mol-1, respectively. All these intra-molecular hyper-conjugative interactions result in intra-molecular charge transfer (ICT) causing stabilization of the system.40
3.5. Chemical Stability and Electronic Transitions
3.5.1. Frontier Molecular Orbitals (FMOs)
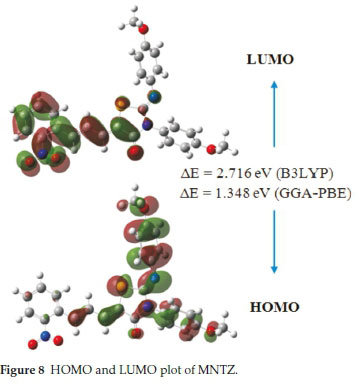
The Frontier molecular orbitals (HOMO and LUMO) play a crucial role in the chemical stability and optical properties of the molecule, as well as in quantum chemistry and UV-Vis spec-trum.41,42 The HOMO, LUMO and band gap energies calculated at B3LYP and PBE functionals with 6-31G(d,p) basis set are listed in Table 2. The distributions of the HOMO and LUMO orbitals computed at the B3LYP/6-31G(d,p) method are shown in Fig. 8. It is interesting to see that both orbitals are substantially distributed over the conjugation plane. As can be seen from Fig. 8, the HOMO orbital is delocalized over the two methoxy-phenyl and imino-thiazolidinone rings whereas the LUMO orbital is localized on the nitro-phenyl and allylidene fragment. This intra-molecular charge transfer from the electron donating group through the π-conjugation system to the electron accepting group promotes the molecular stability.
3.5.2. Global Chemical Reactivity Descriptors (GCRD)
Nowadays, the chemical hardness and softness properties of a molecule are used to describe the reactivity and molecular stability. This relationship can be achieved by the determination of global chemical reactivity descriptor (GCRD) parameters. Using computational methods, GCRD parameters were calculated at the B3LYP and PBE level of theory with 6-31G(d,p) basis set by using the following equations:
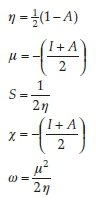
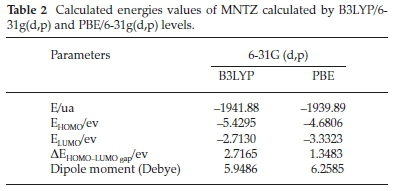
where I = -EHOMO and A = -ELUMO are the ionization potential and electron affinity, respectively. The computed values of GCRD parameters are summarized in Table 3. The chemical hardness (η) values for the title compound are 1.358 and 0.674 eV as obtained by B3LYP and PBE functionals, respectively, where the small ETA-ETA-ETA values indicate that the charge transfer occurs in the molecule.
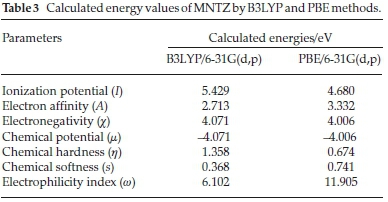
From the results, the calculated ionization potential, electron affinity and electronegativity using B3LYP (PBE) functional are 5.429 (4.680), 2.713 (3.332) and 4.071 (4.006) eV, respectively.
According to these quantum chemical parameters, the MNTZ is a chemically hard system and can be described as a less reactive compound. Furthermore, the high values of the electrophilicity index (6.102 eVbyB3LYP and 11.905 eVby PBE) compared to the low values of chemical potential (-L071 eV by B3LYP and -1:.006 eV by PBE) for the MNTZ prove its electrophilic character. The chemical potential (μ) negative values indicate also the molecular stability of MNTZ. Softness is one of the material properties that measures the extent of chemical reactivity. The MNTZ has low chemical softness values 0.368 eV (B3LYP) and 0.741 eV (PBE), which measure the degree of chemical reactivity.
3.5.3. Absorption Spectra
For a molecule to have an important molecular charge transfer, it must show good absorption and emission properties. TD-DFT is often used to compute the excited-state properties of mole-cules.43,44 From the optimized structure obtained by B3LYP/6-31G(d,p) level of the title compound, the electronic transitions were investigated by using the TD-DFT. The obtained results (vertical excitation energies, oscillator strength (f), transition wavelength and contributions) are collected in Table 4.
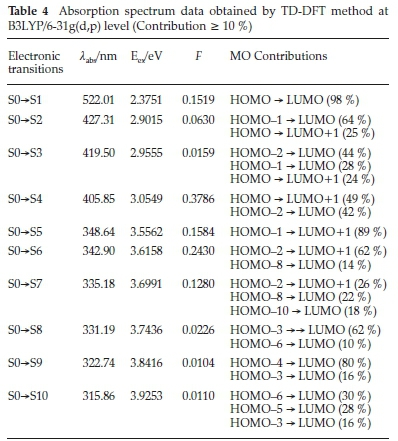
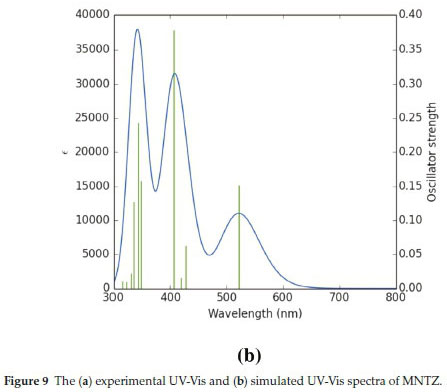
The experimental and calculated UV-Visible absorption spectra are shown in Fig. 9. As can be seen from Fig. 9a, there are two peaks observed at 348 and 406 nm, which are in the ultraviolet region. The corresponding calculated absorption peaks have been found at λ = 342.90 nm, f = 0.2430 and λ = 405.85 nm, f = 0.378. The other important transition in the calculated UV-Visible spectrum (Fig. 9b) is found at λ = 522.01 nm, f = 0.1519 which is in the visible region and corresponds to the HOMO -» LUMO (98 %) transition. The transition simulated at 405.85 nm is comprised of HOMO - LUMO + 1 (49 %) and HOMO-2 -LUMO (42 %) transitions. The electronic excitation contributions from HOMO-8 and HOMO-2 to LUMO and LUMO+1 contribute to the transition which occurs at 342.9 nm. The absorption wavelength at 522 nm attributed to the intramolecular charge transfer (ICT) excitation between donor and acceptor moieties. According to the TD-DFT calculations, the experimental bands at 411 and 356 nm corresponding to π-π* and n-π* transitions. There is a good agreement between experimental and theoretical results.

3.6. Molecular Properties
3.6.1. Mulliken and NBO Atomic Charges
Atomic charge calculations play an important role in the application of quantum chemical calculations to the molecular system. The calculated Mulliken45 and NBO charge values using B3LYP and PBE functionals with 6-31G(d,p) basis set are listed in Table 5. As can be seen, the results of Table 5 reveal the effect of the functional in the value of atomic charge distribution but the same behavior is observed. In fact, for hydrogen atoms, the difference is about 0.01 e and for the non-hydrogen atoms, it does not exceed 0.04 e. The Mulliken atomic charge analysis of MNTZ shows that nitrogen atoms (N2 and N3) have maximum negative charge values which impose positive charges to all carbon atoms bonded to these high electronegative atoms, while the nitrogen atom (N1) posses positive charge which was imposed by oxygen atoms (O1 and O2). The NBO atomic charge analysis of MNTZ shows that oxygen atoms (O3, O4 and O5) have maximum negative charge values. On the other hand, the maximum positive charge value is obtained for the C11 atom (for both Mulliken and NBO charges) due to the negative charge of oxygen (O3). Furthermore, O4 and O5 atoms have high negative charges which impose positive charges to C16 and C22, respectively. Moreover, all the hydrogen atoms have net positive charges thereby all carbon atoms connected to these electropositive atoms exhibit negative charges.
3.6.2. Molecular Electrostatic Potential
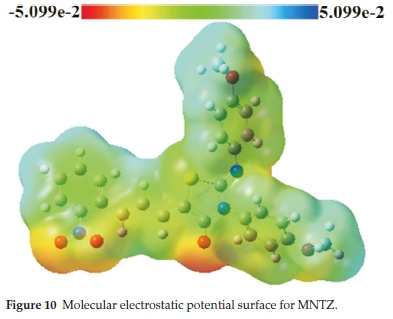
The main purpose of the molecular electrostatic potential study is the localization of electrophilic and nucleophilic sites of a molecule.46 In the MEP map, the red colour indicates the maximum negative region promoting the site for an electro-philic attack while the blue colour indicates the maximum positive region making the site favorable for nucleophilic attack.
Molecular electrostatic potential of MNTZ using B3LYP/6-31G (d,p) optimized geometry is computed and its surface map is shown in Fig. 10. This MEP map shows that there are three possible sites for electrophilic attack localized on the O1, O2, and O3 atoms, while the sites for the nucleophilic attack are located over the hydrogen atoms. The region very near to the sulfur atom is positive due to the fact that the S atom is surrounded by the electropositive atoms. These results show that the most reactive site of the MNTZ molecule is the site containing the oxygen atoms. These sites give information concerning the region from where the compound can have intermolecular interactions.
3.7. Thermodynamic Properties
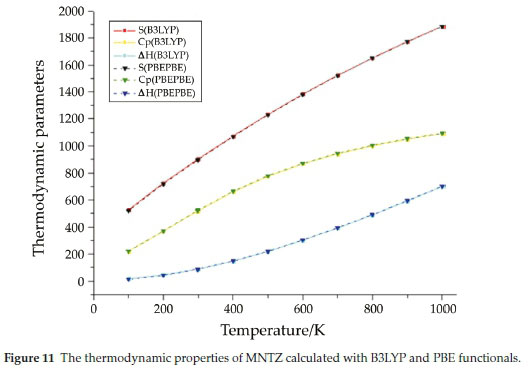
The values of some thermodynamic parameters (such as zero-point vibrational energy (ZPVE), rotational constants, rotational temperatures, thermal energy, molecular capacity at constant volume, entropy, zero point correction, thermal correction to energy, thermal correction to enthalpy and thermal correction to Gibbs Free Energy) of MNTZ at 298.15 K in ground state are obtained from the theoretical harmonic frequencies and reported in Table S7. The variation in zero-point vibrational energies (ZPVEs) seems to be considerable. The ZPVE obtained from B3LYP functional is higher compared to the value obtained from the PBE functional. Whereas, no change was observed for rotational constant and rotational temperature using both functionals. The total energy of the MNTZ molecule is the sum of electronic, translational, rotational and vibrational energies, even for entropy and capacity at constant volume. The thermo-dynamic functions such as entropy (S), heat capacity at constant pressure (Cp) and enthalpy content (ΔΗ = H(T) - H(0)) for various ranges of temperatures from 100 to 1000 K are determined using perl script THERMO.PL47 and reported in Table S8 and Fig. 11. It is obvious to see that the change of the functional does not affect the thermodynamic properties at different temperatures. Furthermore, all the calculated thermodynamic parameters (S, Cp, and increase with the increase of temperature, due to the enhancement of molecular vibration.
4. Conclusions
In the present work, the optimized molecular geometry of thiazolidinones derivative has been investigated and compared with its X-ray structure. The structure was optimized using DFT/B3LYP and PBE methods with 6-31g(d,p) basis set. Bond lengths, as described earlier, are similar to literature values. Bond angles indicate that the π electrons in the studied molecule are delocalized. Except for some values, the obtained results indicate the good agreement between calculated values and experimental parameters. Detailed experimental spectroscopic results were presented. The vibrational wavenumbers of the title compound have been calculated by B3LYP and PBE functionals with 6-31G(d,p) basis set and assigned on the basis of PED. The experimental and theoretical frequencies were compared taking into account the scaling factors 0.961 and 0.986 for B3LYP and PBE, respectively. Gauge independent atomic orbital (GIAO) was used to calculate 1H and 13C NMR spectra. The results showed that, in general, the calculated and experimental chemical shift values were consistent. Hyperconjugative interaction and charge transfer within the molecule were explained by NBO analysis which indicates that the most intra-molecular interactions are due to the overlap between lone pair of O, N and S atoms and antibonding of (C-C), (C-N), (O-N) and (C-O) orbitals. The computed HOMO-LUMO energy gap and the global chemical reactivity descriptors quantum parameters explain the significant charge transfer interactions taking place within the molecule and promotes the molecular stability. Mulliken charges and MEP analysis allow the identification of electrophilic and nucleophilic sites in the molecule and give information about intermolecular interaction regions. Furthermore, the thermodynamic properties of the compound have been calculated and their values at different temperatures are also obtained.
Supplementary Material
Supplementary information is provided in the online supplement.
ORCID iD
R. Rahmani: orcid.org/0000-0002-0335-8783
References
1 M.A. Badawy, N.H. Metwally and D.S. Okpy, Synthesis of some new 5-substituted-3-phenyl-4-thioxo-2-thiazolidinones and their fused thiopyrano[2,3-d]thiazole derivatives, J. Sulfur Chem., 2015, 36(5), 511-525. [ Links ]
2 S.K. Patil, B.P. Langi and H.P. Deokar, Synthesis and characterization of some novel bioactive thiazolidinone derivatives of 3-substituted coumarin, Ind. Am. J. Pharm. Res., 2015, 5(1), 578-583. [ Links ]
3 R. Gupta, Recent advances in chemistry of condensed 4-thiazolidi-nones. J. Heterocyclic Chem., 2016, 53, 1687-1696. [ Links ]
4 E. Hamade, A. Habib, A. Hachem, A.H. Hussein, M. Abbas, T. Hirz, M. Al-Masri and W.H. Faour, Biological and anti-inflammatory evaluation of two thiazole compounds in RAW cell line: potential cyclooxygenase-2 specific inhibitors, Med. Chem., 2012,8(3), 401-08. [ Links ]
5 A. Nowaczyk, M. Kowiel, A. Gzella, L. Fijalkowski, V. Horishny and R. Lesyk, Conformational space and vibrational spectra of 2-[(2,4-dimethoxyphenyl)amino]-1,3-thiazolidin-4-one, J. Mol. Model., 2014, 20, 2366-2375. [ Links ]
6 A. Kunzler, P.D. Neuenfeldt, A.M. das Neves, C.M.P. Pereira, G.H. Marques, P.S. Nascente, M.H.V. Fernandes, S.O. Hübner and W Cunico, Synthesis, antifungal and cytotoxic activities of 2-aryl-3-((piperidin-1-yl)ethyl)thiazolidinones, Eur. J. Med. Chem., 2013, 64, 74-80. [ Links ]
7 M. Asif, Pharmacologically potentials of different substituted coumarin derivatives, Chem. Int., 2015,1(1), 1-11. [ Links ]
8 D. Gautam and R.P. Chaudhary, Synthesis, structure and antimicrobial evaluation of new 3,3a,4,5-tetrahydro-2H-benzo[g]inda-zol-2-yl-thiazol-4(5H)-ones, Spectrochim. Acta A., 2015,135, 219-226. [ Links ]
9 V. Smokal, B. Derkowska, R. Czaplicki, O. Krupka, A. Kolendo and B. Sahraoui, Nonlinear optical properties of thiazolidinone derivatives, Opt. Mater., 2009, 31(3), 554-557. [ Links ]
10 A.S. Yapi, L. Toumi, Y. Lare, G.M. Soto, L. Cattin, K. Toubal, A. Djafri, M. Morsli, A. Khelil, M.A. Del Valle and J.-C. Bernède, On the influence of the exciton-blocking layer on the organic multilayer cells properties, Eur. Phys. J. Appl. Phys., 2010, 50(3), 30403:1-30403:8. [ Links ]
11 R. Rahmani, A. Djafri, J.-C. Daran, A. Djafri, A. Chouaih and F. Hamzaoui, Crystal structure of (2Z,5Z)-3-(4-methoxyphenyl)-2-[(4-methoxyphenyl) imino]-5-[(E)-3-(2-nitrophenyl)allylidene]-1,3-thiazolidin-4-one, Acta Cryst. E, 2016, 72, 155-157. [ Links ]
12 R. Rahmani, A. Djafri, A. Chouaih, A. Djafri, F. Hamzaoui, R. Rizzi and A. Altomare, Synthesis, molecular and solid-state structure of 5-(5-nitrofuran-2-ylmethylen), 3-N-(2-methoxy phenyl), 2-N'-(2-methoxyphenyl) imino thiazolidin-4-one: X-ray powder diffraction and DFT studies, J. Mol. Struct., 2017, 1143, 259-264. [ Links ]
13 N. Khelloul, K. Toubal, N. Benhalima, R. Rahmani, A. Chouaih, A. Djafri and F. Hamzaoui, Crystal structure, Hirshfeld surface analysis and computational studies of thiazolidin-4-one derivative: (Z)-5-(4-Chlorobenzylidene)-3-(2-ethoxyphenyl)-2-thioxothiazolidin-4-one, Acta Chim. Slov., 2016, 63, 619-626. [ Links ]
14 K. Toubal, A. Djafri, A. Chouaih and A. Talbi, Synthesis and structural determination of novel 5-arylidene-3-N(2-alkyloxyaryl)-2-thioxo-thiazolidin-4-ones, Molecules, 2012,17, 3501-3509. [ Links ]
15 N. Benhalima, K. Toubal, A. Chouaih, G. Chita, S. Maggi, A. Djafri and F. Hamzaoui, Synthesis and molecular structure investigation by DFT and X-ray diffraction of ARNO, J. Chem. Crystallogr., 2011, 41, 1729-1736. [ Links ]
16 M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, Jr, J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene,J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, E.O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski and D.J. Fox, Gaussian 09, Gaussian Inc, Wallingford, CT, 2009. [ Links ]
17 A. Frisch, A.B. Nielson and A.J. Holder, GaussView User Manual. Gaussian Inc, Pittsburgh, 2000. [ Links ]
18 A.D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys, 1993, 98, 5648-5652. [ Links ]
19 C. Lee, W. Yang and R.G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B., 1988, 37, 785-789. [ Links ]
20 J.P Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple [Phys. Rev. Lett. 77,3865 (1996)], Phys. Rev. Lett, 1997, 78, 1396-1396. [ Links ]
21 M.H. Jamroz, Vibrational Energy Distribution Analysis VEDA 4, Warsaw, 2004. [ Links ]
22 J.B. Foresman, M. Head-Gordon, J.A. Pople and M.J. Frisch, Toward a systematic molecular orbital theory for excited states, J. Phys. Chem., 1992, 96(1), 135-149. [ Links ]
23 P. Pulay, G. Fogarasi, G. Pongor, J.E. Boggs and A. Vargha, Combination of theoretical ab initio and experimental information to obtain reliable harmonic force constants. Scaled quantum mechanical (QM) force fields for glyoxal, acrolein, butadiene, formaldehyde, and ethylene, J. Am. Chem. Soc., 1983, 105(24), 7037-7047. [ Links ]
24 NIST Computational Chemistry Comparison and Benchmark Database NIST Standard Reference Database Number 101 Release 16a, Editor: Russell D. Johnson III, 2013, http://cccbdb.nist.gov/ [ Links ]
25 R.A. Nyquist, Interpreting Infrared, Raman, and Nuclear Magnetic Resonance Spectra, Volume 1, Academic Press, 2001. [ Links ]
26 G. Socrates, Infrared and Raman characteristic group frequencies tables and charts, 3rd edn., Wiley, 2001. [ Links ]
27 S. Renuga, M. Karthikesan and S. Muthu, FTIR and Raman spectra, electronic spectra and normal coordinate analysis of N,N-dimethyl-3-phenyl-3-pyridin-2-yl-propan-1-amine by DFT method, Spectrochim. Acta A: Mol. Biomol. Spectrosc, 2014,127, 439^53. [ Links ]
28 M. Margoshes and VA. Fassel, The infrared spectra of aromatic compounds: I. The out-of-plane C-H bending vibrations in the region 625-900 cm-1, Spectrochim. Acta, 1955, 7, 14-24. [ Links ]
29 B.H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, Wiley, Chichester, 2004. [ Links ]
30 Shamsuzzaman, K.A.A. Abdul Baqi, A. Ali, M. Asif, A. Mashrai, H. Khanam, A. Sherwani, Z. Yaseen and M. Owais, Synthesis, characterization, biological evaluation and molecular docking of steroidal spirothiazolidinones, J. Mol. Struct, 2015,1085, 104-114. [ Links ]
31 V. Balachandran, G. Santhi, V. Karpagam, B. Revathi and M. Karabacak, Spectroscopic investigation, natural bond orbital analysis, HOMO-LUMO and thermodynamic functions of 2-tert-butyl-5-methyl anisole using DFT (B3LYP) calculations, Spectrochim. Acta A: Mol. Biomol. Spectrosc., 2015,136(Part B), 451-63. [ Links ]
32 P. Mehta, P. Dawedra, V. Goswami and H.S. Joshi, Synthesis and characterization of some thiazolidinone derivatives possessing benzimidazole nucleus, Int. Lett. Chem. Phys. Astron. 2014, 30, 1-8. [ Links ]
33 M. Wolszleger, C.D. Stan, A. Pânzariu, A. Jitäreanu and L. Profire, New thiazolidine-4-ones of ferulic acid with antioxidant potential, Farmácia., 2015, 63(1), 150-154. [ Links ]
34 K. Wolinski, J.F. Hilton and P. Pulay, Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations, J. Am. Chem. Soc., 1990,112(23), 8251-8260. [ Links ]
35 R. Rahmani, N. Boukabcha, A. Chouaih, F. Hamzaoui and S. Goumri-Said, On the molecular structure, vibrational spectra, HOMO-LUMO, molecular electrostatic potential, UV-Vis, firstorder hyperpolarizability, and thermodynamic investigations of 3-(4-chlorophenyl)-1-(1yridine-3-yl) prop-2-en-1-one by quantum chemistry calculations, J. Mol. Struct., 2018,1155, 484-95. [ Links ]
36 J.A. Bohmann, F. Weinhold and T.C. Farrar, Natural chemical shielding analysis of nuclear magnetic resonance shielding tensors from gauge-including atomic orbital calculations, J. Chem. Phys., 1997,107, 1173-1184. [ Links ]
37 E.D. Glendening, A.E. Reed, J.E. Carpenter and F. Weinhold, NBO3.1 Program Manual, Theoretical Chemistry Institute, University of Wisconsin, Madison, 1996. [ Links ]
38 F. Weinhold and C.R. Landis, Discovering Chemistry with Natural Bond Orbitals, John Wiley & Sons, New Jersey, 2012. [ Links ]
39 Y.B. Shankar Rao, M.V.S. Prasad, N. Udaya Sri and V Veeraiah, Vibra-tional (FT-IR, FT-Raman) and UV-Visible spectroscopic studies, HOMO-LUMO, NBO, NLO and MEP analysis of benzyl (imino (1H-pyrazol-1-yl) methyl) carbamate using DFT calculaions, J. Mol. Struct., 2016,1108, 567-582. [ Links ]
40 N. Issaoui, H. Ghalla, F. Bardak, M. Karabacak, N. Aouled Dlala, H.T. Flakus and B. Oujia, Combined experimental and theoretical studies on the molecular structures, spectroscopy, and inhibitor activity of 3-(2-thienyl)acrylic acid through AIM, NBO, FT-IR, FT-Raman, UV and HOMO-LUMO analyses, and molecular docking, J. Mol. Struct., 2017, 1130, 659-668. [ Links ]
41 S. Saravanan, V. Balachandran and K. Viswanathan, Spectroscopic investigation of 4-nitro-3-(trifluoromethyl)aniline, NBO analysis with 4-nitro-3-(trichloromethyl)aniline and 4-nitro-3-(tribromo-methyl)aniline, Spectrochim. Acta A: Mol. Biomol. Spectrosc., 2014, 121, 685-697. [ Links ]
42 I. Fleming, Frontier Orbitals, Organic Chemical Reactions, Wiley, London, 1976. [ Links ]
43 F. Furche and D. Rappoport, in Density functional methods for excited states' equilibrium structure and electronic spectra, in Computational Photochemistry Theoretical and Computational Chemistry, vol. 16, (M. Olivucci, ed.), Elsevier, Amsterdam, 2005. [ Links ]
44 M.A.L. Marques, N.T. Maitra, F.M.S. Nogueira, E.K.U. Gross and A. Rubio, Fundamentals of Time-dependent Density Functional Theory, Springer-Verlag, Berlin Heidelberg, 2012. [ Links ]
45 R.S. Mulliken, Electronic population analysis on LCAOMO molecular wave functions. I, J. Chem. Phys., 1955, 23, 1833-1840. [ Links ]
46 E. Scrocco and J. Tomasi, Electronic molecular structure, reactivity and intermolecular forces: An euristic interpretation by means of electrostatic molecular potentials, Adv. Quantum Chem., 1978, 11, 115-193. [ Links ]
47 K.K. Irikura, THERMO.PL, National Institute of Standards and Technology, Gaithersburg, MD, 2002. [ Links ]
Received 6 February 2019
Revised 9 July 2019
Accepted 9 July 2019
* To whom correspondence should be addressed. E-mail: achouaih@gmail.com
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}