










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.72 Durban 2019
http://dx.doi.org/10.17159/0379-4350/2019/v72a1
RESEARCH ARTICLE
Kinetics and Mechanism of Oxidation of N-acetyl-d-penicillamine in Acidified lodate and Aqueous Iodine
Aqueous IodineI; Kudzanai ChipisoI; Trang DucII; Reuben H. SimoyiI, II, *
IDepartment of Chemistry, Portland State University, Portland, OR 97207-0751, USA
IISchool of Chemistry and Physics, University of KwaZulu-Natal, Westville Campus, Durban, 4000, South Africa
ABSTRACT
The oxidation of the biologically-active thiol, N-acetyl-d-penicillamine (NDPen) by acidified iodate and aqueous iodine has been studied. The stoichiometry of the reaction is 1:1 in which the thiol (RSH) is oxidized to its sulfonic acid (RSO3H) without cleavage of the C-S bond. In excess acidified iodate the reaction displayed a short induction period, followed by formation of aqueous iodine. Overall stoichiometry in excess iodate was 6:5:6IO3- + 5RSH + 6H+ ® 5 RSO3H + 3I2(aq) + 3 H2O. The direct reaction of aqueous iodine and was relatively fast, over within 100 ms and had a stoichiometry of 1:3:3 I2(aq) + RSH + 3H2O ® RSO3H + 6 I- + 6H+. The reaction is essentially diffusion-controlled and our stopped-flow with a mixing time limitation of 1.00 ms could only catch the reaction of the last iodine molecule. This reaction is, however, strongly inhibited by the product of reaction, I-. This is due to the formation of the relatively inert triiodide anion: I2(aq) + I- ® I3-. Mass spectrometry results showed that the reaction proceeds through the sulfinic acid with negligible stabilization of the sulfenic acid. In excess of reductant, the dimeric species is the favoured product due to a rapid condensation-type reaction between the electrophilic unstable sulfenic acid and unreacted thiol.
Keywords: Biological thiols, bioactivation, oxidations, oxyhalogen chemistry.
1. Introduction
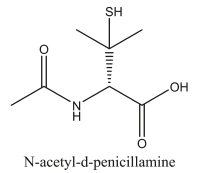
N-acetyl-d-penicillamine (NDPen), an acetylated analog of d-penicillamine (DPen), is one of the chelating agents used clinically as an antidote to metal toxicity.1,2 Heavy metals are ubiquitous in the environment and are associated with serious health effects which include neurological diseases such as Alzheimer's and Parkinson disease.3-5 Chelation therapy is one of the methods in clinical use to reduce those toxic effects.6,7 Some people receive exposure to mercury (Hg) from dental amalgam fillings, although the toxicological consequences of such exposure are still debatable.8,9 NDPen has been recommended and used in the treatment of mercury poisoning as it was found to be more effective than D-penicillamine (DPen) and other agents in mercury extraction.2,10 Chelating agents act by selectively binding toxic metals which are then excreted from the body11. However, some chelating agents are toxic and this limits their use and effectiveness12. For example, the toxicity of Dpen is characterized by alterations in dermal elastic tissue, hypersensi-tivity reactions such as urticarial and morbilliform, as well as lupus-like autoimmune reactions, which are not associated with NDPen.13 Apart from chelation therapy, NDPen has also been used in the treatment of cystinuria, a condition in which there is excess cysteine resulting in painful cystine stones in the bladder and kidney.14 NDPen also reacts with the free cysteine to form a mixed disulfide. This N-acetyl-d-penicillamine-cysteine disulfide is highly soluble in water; about 500 times that of cystine and 10 times that of D-penicillamine-cystine.15 Thus, it makes NDPen more effective in extracting free cysteine. Reactions of NDPen and acidified nitrites to form the thionitrite (SNAP) have been extensively studied and reported in literarure.16,17 S-nitrosothiols have found relevance in biology of vascular homeostasis, neurotransmission and inflammation.18 S-nitroso-N-acetyl-penicillamine (SNAP) generates nitric oxide (NO) spontaneously, thus, it is an important NO donor and is involved in vasodilation of veins and arteries, along with inhibition of platelet aggregation.19 There are other metabolic reactions that take place in the complex physiological environment which can also reduce bioavailability of therapeutic agents.20 Thiols, for example that of DPen, are easily oxidized to a disulfide. Though minor, the extent to which such side-reactions take place may alter the therapeutic value of the chelating agents. Albeit in low concentrations, oxyhalogen species such as hypohalous acids; HOCl, HOBr and HOI, which are present in the physiological environment, have been observed to oxidize thiols. The kinetics and mechanism of oxidation of NDpen by acidified iodate and aqueous iodine is reported here.
2. Experimental Procedures
2.1. Materials
The following reagent grade chemicals were used without further purification: sodium iodate, perchloric acid (70-72 %), sodium iodide, iodine, sodium perchlorate, soluble starch, sodium thiosulfate (Fisher) and N-acetyl-d-penicillamine (Sigma). Iodine solutions, being volatile, were kept capped and standardized spectrophotometrically before each set of experiments. Stock solutions of N-acetyl-d-penicillamine were prepared just before use.
2.2. Methods
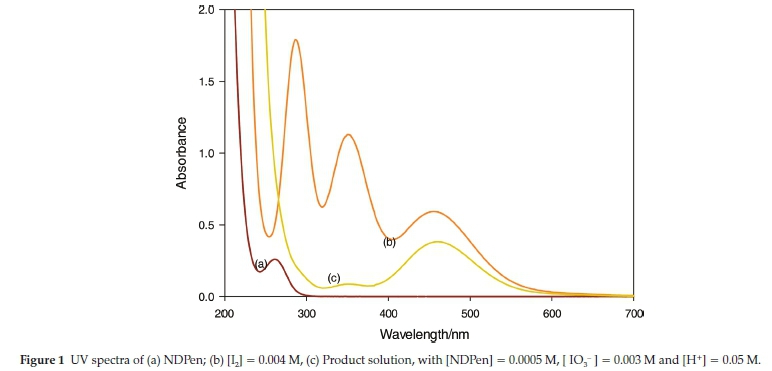
The rapid reactions of NDPen with iodine were followed on a Hi-Tech Scientific™ SF61-DX2 double-mixing stopped-flow spectrophotometer. These reactions were monitored by following consumption of iodine at 460 nm (e = 770 M-1 cm-1). NDPen has no absorbance in the visible region, while aqueous iodine has an isolated peak at 460 nm, which is its isosbestic point with triiodide, I3- (see Fig. 1). Thus, absorbance at this peak was used for analytical determination of aqueous iodine. Slower reactions involving oxidation of NDPen by iodate were monitored on a conventional Perkin-Elmer Lambda 25 UV-Vis spectrophotometer. All kinetics experiments were performed at 25.0 ± 0.1 °C and at an ionic strength of 1.00 M (NaClO4). All solutions were prepared with doubly-distilled deionized water from a Barnstead Sybron Corporation water purification unit capable of producing both distilled and deionized water (Nanopure). Mass spectra of product solutions were taken on a Thermo Scientific LTQ-Orbitrap Discovery mass spectrometer (San Jose, CA) equipped with an electrospray ionization source operated in the positive mode.
2.2.1. Stoichiometric Determinations
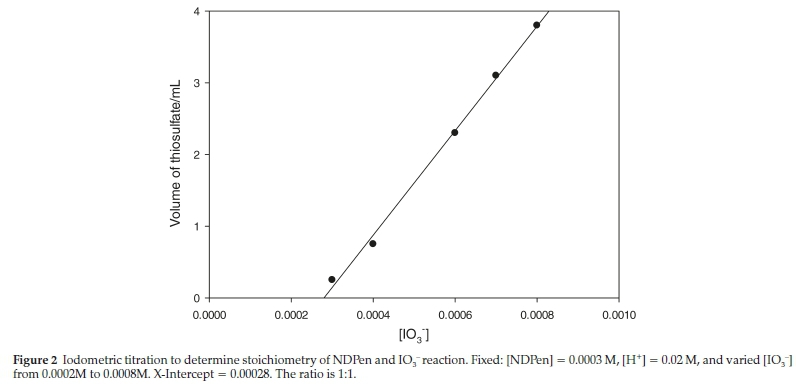
The stoichiometry of the reaction was determined both spectrophotometrically and titrimetrically. NDPen has no measurable absorbance in the visible region (spectrum (a), Fig. 1). Aqueous iodine has an isosbestic point with triiodide at 460 nm. It has other peaks at 353 nm and 286 nm (spectrum (b)), but these were not isolated, and could not be used for rate nor stoichiometric determinations. For a fixed concentration of acidified NDPen (0.30 mM), a series of varying concentrations of excess iodate were added and solutions left to stand overnight. The excess oxidizing power left after total consumption of NDPen was determined iodometrically by adding excess iodide and titrating the liberated iodine against standard thiosulfate with starch as indicator.21
The volume of thiosulfate was then plotted against initial iodate concentrations (see Fig. 2). The linear plot is then extrapolated to the iodate axis to derive the concentration of iodate needed to just oxidize NDpen with no iodate left to produce iodine from the Dushman reaction.22 Figure 2 shows an intercept value of 0.29 mM iodate which represents the stoichiometric amount of iodate needed to consume 0.30 mM NDPen. This suggests a 1:1 stoichiometry of iodate to NDPen in which the NDPen loses six electrons, and oxidation occurs only on the sulfur centre to give a sulfonic acid, NDPen-SO3H:
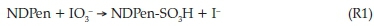
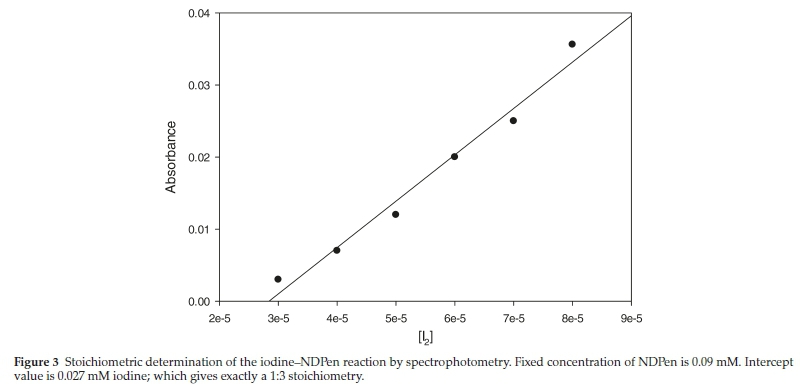
NMR analysis had also shown that the carbon skeleton is left unchanged, the only difference being the downfield shift of all the protons due to the strong electron-withdrawing effect of the sulfonic acid group. The asymmetric carbon centre is left untouched in this reaction since the oxidation occurs distal to this centre. The stoichiometric determination of the direct reaction of iodine and NDPen was performed both titrimetrically and spectrophotometrically. In the spectrophotometric analysis, excess iodine concentrations were used over a fixed concentration of the substrate NDPen of 0.09 mM. For each experiment, the final absorbance was noted. These experiments were performed in triplicate. This final absorbance concentration was plotted against the initial iodine concentrations and extrapolated to zero absorbance. This is the iodine concentration needed to just consume 0.09 mM NDPen. Figure 3 shows an intercept value of 0.027 mM iodine, which strongly suggests a 1:3 stoichiometric ratio, with the oxidation of NDPen only as far as the sulfonic acid as was observed with the iodate oxidation:
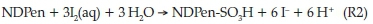
Excess acidic iodate solutions gave iodine as the final product of oxidation. In highly excess iodate conditions, iodine formation was determined by the initial NDPen concentrations. In excess acidic iodate conditions, after reaction R1 is satisfied and all the substrate is fully oxidized, the Dushman reaction22 (R3) takes over to consume all the iodine formed in reaction R1:
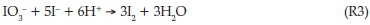
Thus, a linear combination of 5R1 + R3 removes iodide as a product, and replaces it with aqueous iodine for an overall stoichiometry of:
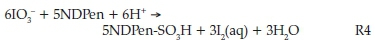
Thus, the amount of iodine produced was determined by the initial substrate concentrations. Specifically, in high excess of acidic iodate, the aqueous iodine produced was exactly 60 % of the initial concentration of NDPen.
2.2.2. Product Determination
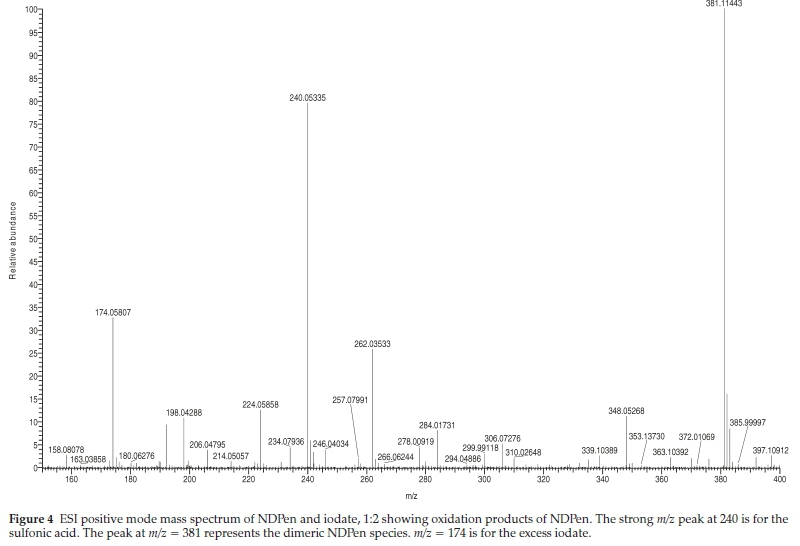
The product solution was treated with barium chloride. No barium sulfate precipitate was observed; indicating that the carbon-sulfur bond in NDPen was not cleaved during the oxidation. Figure 4 shows the ESI-MS spectrum of the product of a 1:2 ratio of NDPen to acidic iodate. The spectrum shows a strong peak at m/z = 240.05335 which represents addition of three oxygen atoms to the substrate NDPen, which is expected to show up at m/z = 192.00828. The strong peak at m/z = 381.11443 is the dimeric NDPen species. The peak at 262.03533 is the sodium salt of the sulfonic acid at m/z = 240.05335. There was also a strong peak for the sulfinic acid at m/z = 224.05858. The sulfenic acid was not observed. If formed, it is expected to react with the unoxidized NDPen in a condensation-type reaction to produce the dimer, which is in highest abundance at these conditions. The peak at 174.05807 represents unreacted iodate. A low concentration of acid was deliberately utilized in this reaction to slow it to enable the observation of any intermediates that are formed. At higher acid concentrations the dimeric species is not dominant, and if it is formed, it is slowly further oxidized to the final sulfonic acid product.
2.2.3. Reaction Dynamics
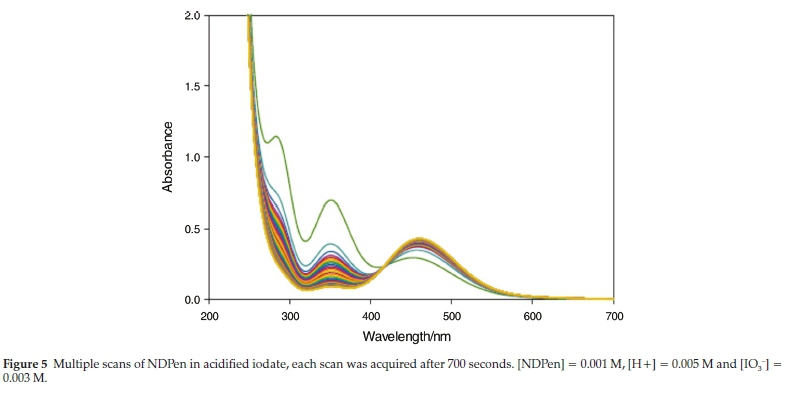
All kinetics experiments reported here were run in excess acidic iodate. In this format, we could utilize formation of iodine to follow the reaction. For iodate oxidations, the reaction was relatively fast; it was over in less than 12 min (attainment of stoichiometry R4) at mild pH conditions of between 2 and 3. At lower pH conditions, the reaction had to be monitored on a stopped-flow ensemble. Figure 5 shows scans of the reaction solutions between 200 and 700 nm. Each scan is taken 700 s apart. The first trace (green), shows peaks at 286 nm, 353 nm and 460 nm. The first two peaks are attributed to the triiodide, I3-, and the last is the known isosbestic point of triiodide with iodine. Progressive scans show a decrease in absorbances at 286 and 353 nm with a monotonic increase in the peak at 460 nm. The first trace, after 700 s, already shows iodine formation. Subsequent traces after this show a monotonic increase in the iodine peak until the maximum absorbance as predicted by R4 is attained.
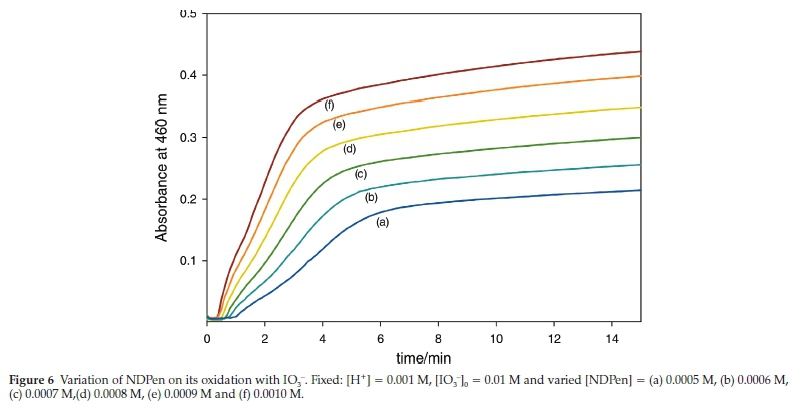
Figure 6 shows the absorbance traces, at fixed iodate and acid concentrations, while varying the initial NDPen concentrations. These traces were all taken at high excess of oxidant, at least a 10:1 ratio. The reaction commences with an induction period in which no iodine production is observed. This induction period is inversely proportional to the concentration of NDPen and the rate of formation of iodine after the induction period is proportional to the initial concentration of NDPen. Most oxyhalogen-sulfur reaction kinetics deliver an almost invariant induction period at these conditions.
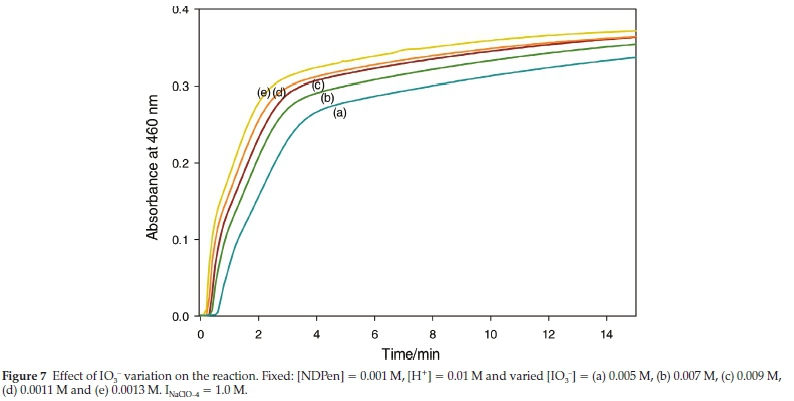
Figure 7 shows the variation of initial iodate concentrations. As usual, they all have to be at stoichiometric excess of iodate for iodine to be produced. In Figure 7, the ratio was varied from 5 to 13. All traces shown in this figure gave the same final iodine absorbance of 0.46. Traces were truncated before full reaction. After expanding the first minute of the reaction, it was noted that there was an inverse relationship between the initial iodate concentrations and the quiescent period before formation of iodine.
Figure 8 shows the effect of acid on the reaction. The oxidant to reductant ratio was fixed at 20 and acid was varied between 1.0 and 3.5 mM. Higher acid concentrations gave shorter induction periods and a faster rate of iodine formation. Within a small range of low acid concentrations, the inverse of the induction periods (proportional to rate of reaction) had a square dependence on the acid concentration. As acid was increased, past 3.0 mM, this linear relationship was lost (figure not shown).
2.2.4. Effect of Iodide
Iodide had a catalytic effect on the reaction. Very small amounts of iodide, 10 micromolar, decreased the induction period and the rate of formation of iodine at the end of the induction period. In higher iodide concentrations and in excess iodate conditions, final iodine formation was enhanced by the additional iodide, which was converted to iodine.
2.2.5. Iodine-NDPen Kinetics
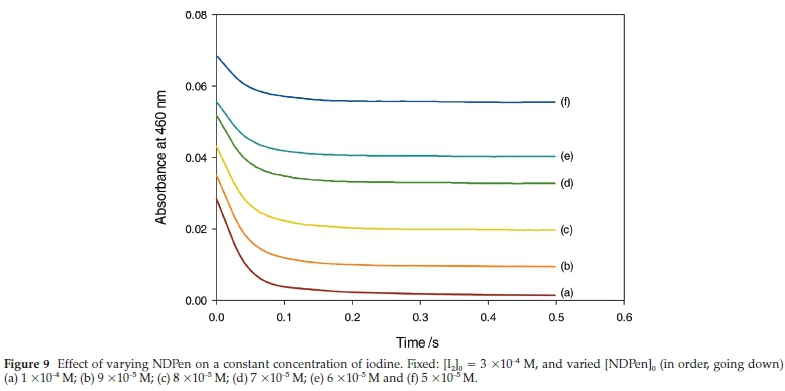
The direct reaction of iodine and NDPen is one of the three important reactions in the overall reaction scheme. Its rate of reaction will determine whether a monotonic increase in iodine concentrations is observed or whether oligooscillations in iodine concentrations, instead, are observed. If it is orders of magnitude faster than the iodate-NDPen reaction, then it should be able to consume all the iodine formed from the Dushman reaction. Thus, observation of iodine would indicate that all the NDPen has been totally consumed. Figure 9 shows the absorbance traces for the iodine-NDPen reaction at constant iodine concentrations while varying NDPen concentrations.
Of note is the fact that the reaction is extremely rapid, essentially over in 200 ms and approaches the limit of observation of our stopped-flow ensemble. It is indeed much faster than the main iodate-NDPen reaction. Further examination of Fig. 9 shows the following: (a) trace (a) is the stoichiometric mixture of 1:3 iodine to NDPen and thus we expect all the iodine to be consumed with a resultant zero absorbance of iodine obtained at the end of the reaction. This is indeed the case. And (b) we expect the starting absorbance of iodine at the beginning of the reaction, for all traces, to be 0.231. The observed starting absorbances of iodine are much lower than the expected 0.231 value, and range from 0.027 to 0.068. This indicates that there is a substantial part of the reaction that is so rapid that it cannot be captured on our stopped-flow instrument, which has a mixing time limitation of 1 ms. If we concentrate on the stoichiometric trace, trace (a), the starting absorbance of 0.027 would suggest that the first two iodine consumption steps are so fast that they are essentially diffusion-controlled, and we can only observe part of the last iodine consumption step. Another series of experiments were undertaken while varying the initial iodine concentrations. They mirror the results shown in Fig. 9 of bimolecular kinetics: the reaction is first order in both iodine and NDPen. Since most of the reaction could not be followed in the stopped-flow, one could only determine a lower-limit bimolecular rate constant of 1.95 x104M-1s-1.
Acid had a very small inhibitory effect on the iodine-NDPen reaction. We had to utilize high acid concentrations, much higher than those utilized in the series of experiments reported in this manuscript, for us to observe any discernible retardation. For the pH ranges used here, pH 2 to 3; acid had no apparent effect on the reaction.
2.2.6. Effect of Iodide
Generally, the reaction starts off rapidly and slows down as it proceeds (see Fig. 10). This would suggest some form of autoinhibition. The product of reduction of iodine is iodide, and so this was used to determine if it, indeed was the autoinhibitory species. Figure 10 shows a series of experiments in which iodide was deliberately added to the iodine-NDPen reaction. This figure shows a dramatic reduction in rate with added iodide, to the extent that the initial absorbances observed are close to those that are expected from a simple calculation, indicating that the stopped-flow ensemble is now able to capture the whole reaction. Iodide reacts with iodine to produce triiodide, which is not as effective an electrophile as iodine:23

All traces shown in Fig. 10 were run in excess of iodide, ensuring nearly quantitative formation of triiodide. For example, in trace (a) of the figure, fully 99.2 % of the iodine is transformed to triiodide. Thus, one can assume the sole oxidant in Fig. 10 is triiodide, and hence the observed sluggish kinetics. The difference in the final effective iodine concentration in all traces in Fig. 10 is minimal, and yet there is a large discernible reaction rate retardation with iodide. No simple relationship could be obtained between rate and iodide concentrations, indicating a multi-term rate law. One can then assume a simple electrophilic attack by iodine on the thiol centre of NDPen, RSH:
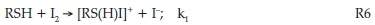
This is then followed by hydrolysis to yield the unstable sulfenic acid which we were unable to detect

The sulfenic acid is highly unstable and electrophilic and will either be oxidized further to the more stable sulfinic acid:

Or in conditions of excess reductant, it will be converted predominantly to the dimeric species via a condensation-type reaction with unreacted thiol (see peak at m/z = 381.1144 in Fig. 4):

In the very extreme case, we can assume that triiodide is inert, i.e. k2is vanishingly small:

We can then derive a rate of consumption of iodine of the form:

The subscript '0' denotes initial concentration. With the high value of Keq, even small values of iodide can induce visible and quantifiable retardation. Thus, as the reaction proceeds, iodide concentrations increase and the reaction autoinhibits.
3. Mechanism
The reaction dynamics suggest that the standard oxyiodine reactions are dominant in this mechanism. The inverse of the induction time strongly correlates with the rate of reaction since production of iodine indicates a specific point in the reaction's progress, i.e. end of reaction stoichiometry R1. The dependence of the reaction rate on the iodate concentrations and on acid concentrations to the second order implicate the well-known first step in the reduction of iodate24:
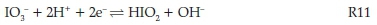
The two electrons can be supplied by any two-electron reductant. This initial step has been extensively studied and its kinetics have been evaluated in a composite form as:

where Red is any two-electron reductant. If Red is iodide, then the rate-limiting step can be written as22:

HIO2 and HOI represent the active oxyiodine species that perform the bulk of the subsequent oxidations. Iodate itself is relatively inert. Thus any induction period observed denotes the time taken for the build-up of the reactive species. Iodate solutions always contain at least 10-6 M of iodide ions, but these are insufficient to initiate the reaction even though only catalytic amounts are needed. The initial iodide production is derived from an initial direct reaction of NDPen with acidic iodate:

The rest of the iodide ions are produced as the reaction proceeds. Further reaction of HOI, for example, immediately produces I- which can be recycled and used in reaction R12:
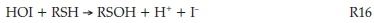
HIO2 can also disproportionate in the presence of iodide to produce more HOI:25

Reactions R12 and R17 have been extensively studied25, and their kinetics parameters are known. If sequence R13 to R16 was the dominant pathway of the oxidation of NDPen, then we would expect first order kinetics in acid. However, this sequence is only an initiation process which introduces iodide to initiate reaction R12, through which the bulk of the oxidation of NDPen occurs, and thus second order kinetics in acid prevails.
3.1. Iodine Formation
There is a single reaction that can be identified in this reaction network as being responsible for the formation of iodine. It is the reverse of the iodine hydrolysis reaction26:
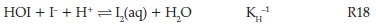
Though it is a composite reaction it is very rapid in the forward direction, with a rate constant of 3.1 x1012 M-2 s-1. Thus, as long as HOI is present, in acidic medium, the reaction will mop up all the iodide as soon as it is formed. This reaction has to compete with the other reactions that consume iodine. The predominant route to consumption of iodine is the direct reaction of iodine with the substrate; reaction R6. Since we have determined it to be much faster than the other reactions that compete for HOI, it means then, formation of iodine indicates complete consumption of NDPen. This simplifies the kinetics and interpretation of the observed induction periods.
Figure 9 shows that this reaction is autoinhibitory, but as long as there is a sink for iodide, which, in this case, is made up of reactions R17 and R18; autoinhibition will not assert itself.
3.2. Overall Reaction Mechanism
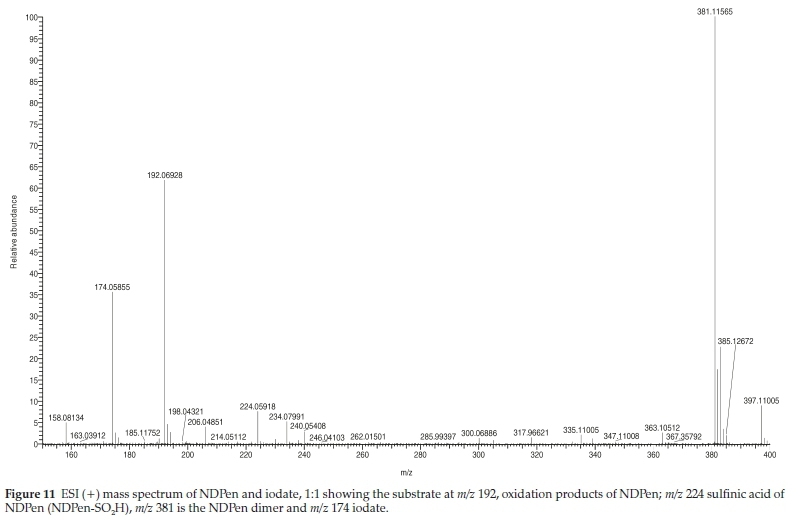
Figure 11 shows the ESI spectrum in the positive mode of a 1:1 mixture of iodate and NDPen (stoichiometric mixture). Acid concentrations were reduced to slow the reaction so that any viable intermediates could be observed. The spectrum was taken before full reaction had occurred. The spectrum shows the expected strong peak for the unreacted NDPen at m/z = 192.06928 and the dimer at 381.11565. There is a relatively strong peak observed for the sulfinic acid at m/z = 224.05918. Since it is an intermediate, its appearance shows that it is stable enough to be observed. No trace is observed of the sulfenic acid, which would have been observed at about 208.08. This is the first intermediate observed, and thus it is rapidly mopped up by unreacted thiol to form a dimer (reaction R9). All iodine formation traces in excess iodate conditions show an initial rapid iodine formation followed by a discontinuity and a slower iodine formation until stoichiometry R4 is satisfied. The rate of iodine formation is dependent on the rate of oxidation of NDPen, which produces the reactive intermediates. After formation of the sulfinic acid, the slower rate of formation of iodine is derived from the slower rate of oxidation of the sulfinic acid.
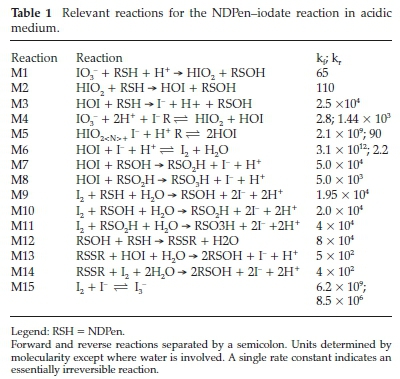
The full mechanism is summarized in Table 1. It is made up of a compact set of 14 reactions. The first three reactions, M1 to M3 are the initiation reactions. They are only valid at the beginning of the reaction to produce the catalytic amounts of iodide needed to assert reaction M4 and its subsequent cascade of M5 and M6. There are three oxidants in the reaction mixture: HIO2 HOI and I2(aq). However, disproportionation reaction M5 is so rapid that HIO2 can be omitted without any loss of accuracy. Reactions M7 to M11 involve the oxidation of the NDPen intermediates to the sulfonic acid product. Reaction M12 is responsible for production of the dimeric NDPen species observed at m/z = 381.11565. Further oxidation of the dimeric species (M14 and M15) is slower than NDPen's oxidation. Lower quantities of the dimeric species are observed in high excess of oxidant, as the sulfenic acid is rapidly oxidized to the more stable sulfinic acid. The autoinhibition is contained in reaction M15. In this network, we assumed that I3- is totally inert as applied in equation (1). Veracity of simulations was difficult to acertain because they could be fitted by adjusting the kinetics parameter for reaction M1, the initial initiation reaction responsible for the 'seed' iodide that is then used in reaction M4 to start the cascade of the well-known oxyiodine reactions. With the fast rate of reaction M9 and subsequently further oxidation of the sulfenic acid, the simulation was effectively dominated by the Dushman reac-tion,22 which was not illuminating on the main reaction under study.
4. Conclusions
This short kinetics and mechanistic study has shown that the oxidation of NDPen proceeds through the formation of the dimeric species which is more stable and less nucleophilic than NDPen itself. As in most biologically-active thiols, the carbon-sulfur bond is not cleaved even under strong oxidizing conditions.
Acknowledgements
This work was supported by a National Science Foundation grant number CHE-1056366 and a partial Research Professor grant from the University of KwaZulu-Natal.
§ ORCID iDs
Τ. Due: orcid.org/0000-0001-8655-5063
R.H. Simoyi: orcid.org/0000-0002-2965-1582
References
1 M. Blanusa, V.M. Varnai, M. Piasek and K. Kostial, Chelators as antidotes of metal toxicity: therapeutic and experimental aspects, Curr. Med. Chem., 2005,12, 2771-2794. [ Links ]
2 R.A.P. Kark, D.C. Poskanze, J.D. Bullock and G. Boylen, Mercury poisoning and its treatment with N-acetyl-D,L-penicillamine, New Engl. J. Med., 1971, 285, 10-14. [ Links ]
3 A. Budimir, Metal ions, Alzheimer's disease and chelation therapy, Acta Pharm., 2011, 61, 1-14. [ Links ]
4 Y. Nishida, The chemical process of oxidative stress by copper(II) and iron(III) ions in several neurodegenerative disorders, Monatsh. Chem., 2011,142, 375-384. [ Links ]
5 H.R. Pohl, N. Roney and H.G. Abadin, Metal ions affecting the neurological system, Met. Ions Life Sci., 2011, 8, 247-262. [ Links ]
6 S.J.S. Flora and V Pachauri, Chelation in metal intoxication, Int. J. Environ. Res. Pub. Health, 2010, 7, 2745-2788. [ Links ]
7 O. Andersen, Principles and recent developments in chelation treatment of metal intoxication, Chem. Rev., 1999, 99, 2683-710. [ Links ]
8 L. Bjorkman, G.Sandborgh-Englund and J.Ekstrand, Mercury in saliva and feces after removal of amalgam fillings, Tox. Appl. Pharmacol., 1997,144, 156-162. [ Links ]
9 J. Ekstrand, L. Bjorkman, C. Edlund and G. Sandborgh-Englund, Toxicological aspects on the release and systemic uptake of mercury from dental amalgam, Eur. J. Oral Sci., 1998,106, 678-686. [ Links ]
10 L. Markowitz and H.H. Schaumburg, Successful treatment of inorganic mercury neurotoxicity with N-acetyl-penicillamine despite an adverse reaction, Neurology, 1980, 30, 1000-1001. [ Links ]
11 S. Acharya, S. Chaudhuri, S. Chatterjee, P. Kumar, Z. Begum, S. Dasgupta, S.J.S. Flora and S. Chaudhuri, Immunological profile of arsenic toxicity: a hint towards arsenic-induced carcinogenesis, Asian Pacific J. Cancer Prevention, 2010,11, 479-90. [ Links ]
12 J.L. Domingo, Developmental toxicity of metal chelating agents, Reproduct. Toxicol., 1998, 12, 499-510. [ Links ]
13 B.K.H. Lewis, P.L. Chern and M.S. Stone, Penicillamine-induced elastosis of the mucosal lip, J. Am. Acad. Dermatol, 2009,60,300-303. [ Links ]
14 O. Traxer, E.Lechevallier and C.Saussine, Cystinuria: diagnosis and management, Progres Urologie, 2008, 18, 832-836. [ Links ]
15 G.S. Stokes, J.T. Potts, M. Lotz and F.C. Bartter, New agent in treatment of cystinuria - N-acetyl-D-penicillamine, Br. Med. J., 1968, 1, 284-288. [ Links ]
16 I. Chipinda and R.H. Simoyi, Formation and stability of a nitric oxide donor: S-nitroso-N-acetylpenicillamine, J. Phys. Chem. B, 2006, 110, 5052-5061. [ Links ]
17 L. Field, R.V Dilts, R. Ravichandran, P.G. Lenhert and G.E. Carnahan, Unusually stable thionitrite from N-acetyl-D,L-penicillamine - X-ray crystal and molecular-structure of 2-(acetylamino)-2-carboxy-1,1-dimethylethyl thionitrite, J. Chem. Soc. Chem. Comm. 1978, 249-250. [ Links ]
18 N. Hogg, Biological chemistry and clinical potential of S-nitroso-thiols, Free Rad. Biol. Med, 2000, 28, 1478-1486. [ Links ]
19 M.R. Miller and I.L. Megson, Review - Recent developments in nitric oxide donor drugs, Br. J. Pharmacol, 2007,151, 305-321. [ Links ]
20 L. Peters, G.B. Steventon, S. Green, S. Sturman, R.H. Waring and A.C.Williams, D-penicillamine metabolism in neurodegenerative diseases - An in-vivo in-vitro sulfhydryl methylation study, Xenobiotica, 1994, 24, 1013-1020. [ Links ]
21 A. Indelli. Kinetic study on the reaction of sodium chlorite with potassium iodide, J. Phys. Chem., 1964, 68, 3027-3031. [ Links ]
22 H.A. Liebhafsky and G.M. Roe. The Dushman reaction. Int. J. Chem. Kinet, 1971,11, 693-701. [ Links ]
23 D.H. Turner, G.W. Flynn, N. Sutin and J.V. Beitz. Laser Raman temperature-jump study of the kinetics of the triiodide equilibrium. J. Am. Chem. Soc., 1972, 94, 1554-1559. [ Links ]
24 G. Schmitz, Kinetics of the Dushman reaction at low I- concentrations, Phys. Chem., Chem. Phys., 2000, 2, 4041-044. [ Links ]
25 J.A. Agreda, R.J. Field and N.J. Lyons, Kinetic evidence for accumulation of stoichiometrically significant amounts of H2I2O3 during the reaction of I- with IO3-, J. Phys. Chem. A, 2000, 104, 5269-5274. [ Links ]
26 K. Kustin and M. Eigen, Disproportionation kinetics of halogens studied by temperature-jump spectrophotometry, J. Am. Chem. Soc., 1962, 84, 1355-1359. [ Links ]
Received 17 January 2018
Revised 26 September 2018
Accepted 26 September 2018
* To whom correspondence should be addressed. E-mail: rsimoyi@pdx.edu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}