










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.71 Durban 2018
http://dx.doi.org/10.17159/0379-4350/2018/v71a18
RESEARCH ARTICLE
Molecular Complexes of Boron Trifluoride with some Formyl Compounds, HCOX (X = H, CH3, NH2, OH, F): Effect of Substitution, and Extension to X = Li, BeH and BH2‡
T. Anthony Ford*
School of Chemistry and Physics, University of KwaZulu-Natal, Westville Campus, Private Bag X54001, Durban, 4000, South Africa
ABSTRACT
The complexes formed between boron trifluoride and formaldehyde, acetaldehyde, formamide, formic acid and formyl fluoride have been studied by means of ab initio calculations at the second order level of M0ller-Plesset perturbation theory (MP2), using Dunning's aug-cc-pVTZ basis sets. The structures, the interaction energies, the perturbations of the vibrational spectra and the charge transfers occurring on complexation, both of the BF3 and of the base moieties, have been determined and the variations in these properties have been rationalized in relation to the nature of the X fragments (X = H, CH3,NH2, OH, F). The calculations have been extended to include the putative complexes between BF3 and the presently unknown compounds HCOLi, HCOBeH and HCOBH2, in order to complete the range of formyl compound bases with heteroatoms across the whole of the first row of the periodic table.
Keywords: Ab initio calculations, boron trifluoride, molecular complexes, structures, energies, vibrational spectra.
1. Introduction
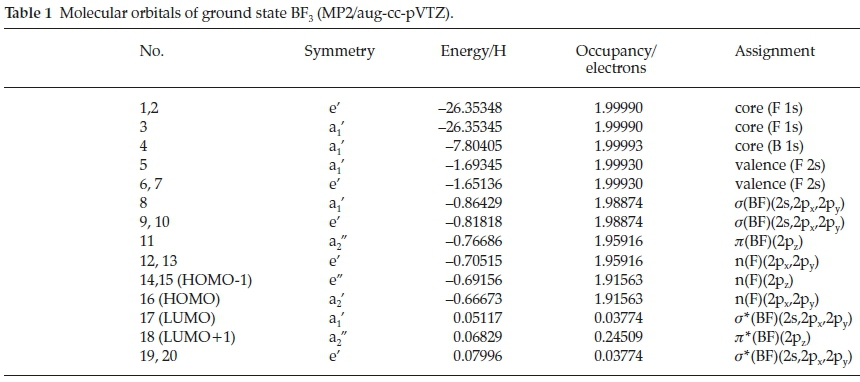
The boron trihalides have long been regarded as prototypical electron acceptors in Lewis acid-base complexes.1-3 This type of interaction is one of a number of non-covalent interactions4 that have received a great deal of attention in recent years, named p-hole bonding5 or triel bonding.6 Since the elucidation by Latimer and Rodebush in 1920 of the nature of the hydrogen bond,7 a series of named types of non-covalent interaction has been recognized. These include the lithium,8 the halogen,9 the chalcogen,10 the beryllium,11 the pnicogen12 and the tetrel bonds.13 These names reflect mainly the group of the periodic table in which the major atom taking part in the interaction is found, and most of these families of interactions can be described by the generic title of ó-hole bonds.14 In the case of boron trifluoride, the origin of the interaction is donation from a lone pair orbital or a p-electron system of the base to the r*(BF3) orbital of the acid. This BF3 orbital is the LUMO+1 orbital of a2" symmetry, as indicated in Table 1 and illustrated in Fig. 1. The region of space occupied by this orbital is a p-hole.5
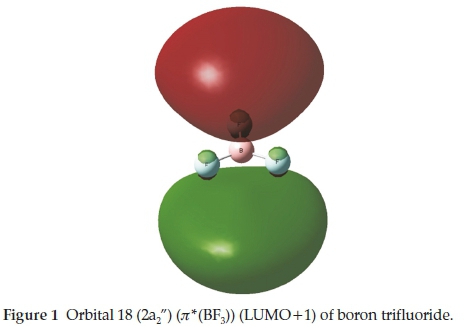
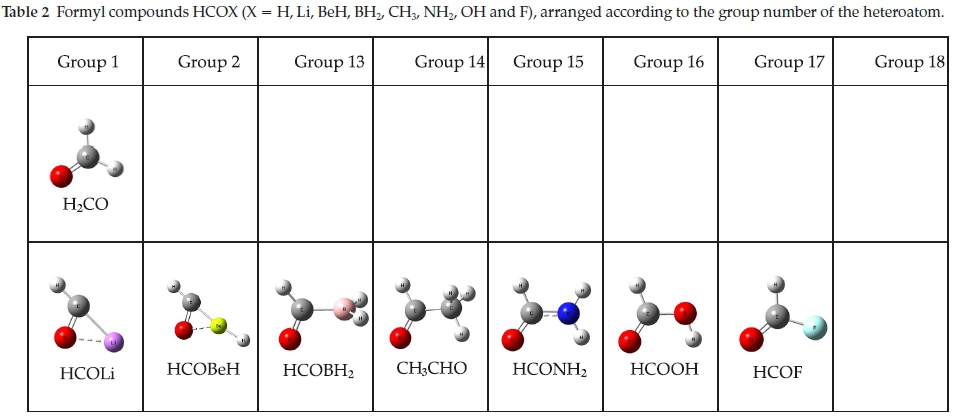
We have reported the results of our ab initio calculations of the properties of the complexes of BF3 with the formyl compounds H2CO, CH3CHO and HCOF, as well as a number of related oxygen, nitrogen and sulphur bases.15 We now extend our list of oxygen bases to include formamide and formic acid, with the aim of determining any trends which may exist in the properties of these adducts which might be related to the position of the heteroatom in the periodic table. Although the molecules HCOLi, HCOBeH and HCOBH2 are not known, it would be instructive to include their tentatively proposed complexes with BF3 in our study, in order to investigate the possible influence of the less common atom and groups, Li, BeH and BH2. The whole series of oxygen bases is represented in Table 2, which also illustrates the optimized structures of the monomers.
2. Computational Methodology
The computations were carried out using the Gaussian 09 program16 at the second order level of M0ller-Plesset perturbation theory (MP2)17 and with the augmented correlation-consistent polarized valence triple-zeta basis sets of Dunning.18 The interaction energies were corrected for basis set superposition error (BSSE),19 using the full counterpoise technique of Boys and Bernardi,20 and for zero-point vibrational energy differences. The vibrational analyses were performed at the harmonic level, and the reported wavenumbers and their shifts are unscaled.
‡This paper was presented at the 20th South African Chemical Institute Inorganic Chemistry Conference, Hermanus, South Africa, 25-29 June 2017.
Natural atomic charges, and their changes on complexation, were computed using the NBO program of Glendening et al?1, which is part of Gaussian 09.16
3. Results and Discussion (X = H, CH3,NH2, OH, F)
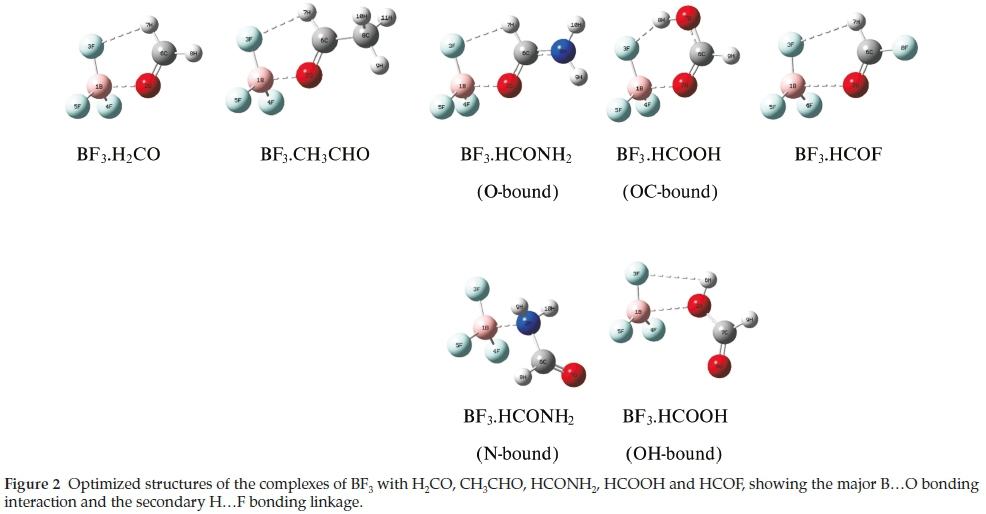
The range of oxygen bases is presented in Table 2, arranged according to the group number of the heteroatom. This table also illustrates the optimized structures of the base monomers. In each case the HCO group of the base was found to be coplanar with one of the BF bonds of BF3, in structures of Cs symmetry. The only exception was the N-bound complex BF3.HCONH2 which optimized in C1 symmetry. Two stable structures were found for the adducts with HCONH2 and HCOOH, with binding occurring between the B atom and the O or N atoms in the first case, and between B and the carbonyl or hydroxyl O atoms in the second. The O-bound structure of BF3.HCONH2 and the OC-bound structure of BF3.HCOOH were found to be the global minima on the respective potential energy surfaces. The Cartesian coordinates of the optimized structures are given in the Supplementary Information, in Tables S1 to S7.
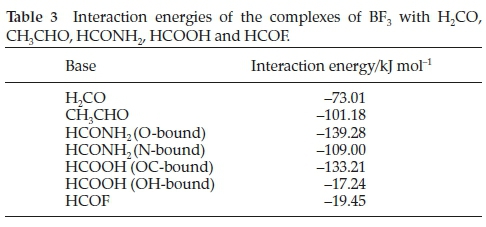
3.1. Interaction Energies
The computed interaction energies are listed in Table 3. The energies change in a non- monotonic manner with respect to the position of the heteroatom in the periodic table, increasing from the H2CO to the HCONH2 species, then diminishing from the HCONH2 to the HCOF complexes.
3.2. Molecular Structures
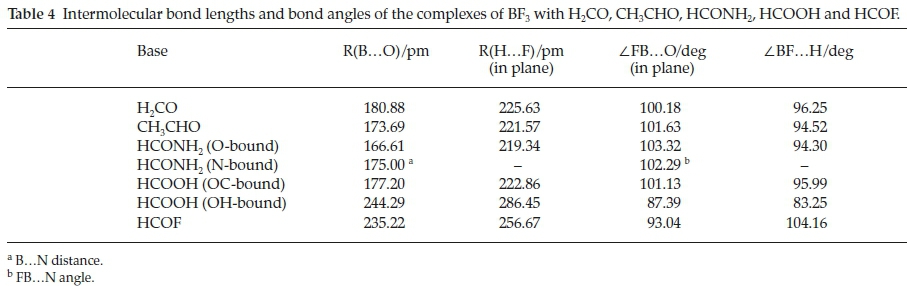
The optimized structures of the seven complexes are presented in Fig. 2. The relevant intermolecular geometrical parameters (the B...O and H...F distances and the in-plane FB...O and BF...H angles) are collected in Table 4. The B...O and H...F distances and the BF.H angles decrease while the FB.O angles increase withincreasinginteractionenergy,correlatingin a regular manner, as shown in Fig. 3. The outlier in each relationship is the BF3.HCOOH complex, in which the F.H interaction occurs through the hydroxyl rather than the formyl hydrogen atom of formic acid.
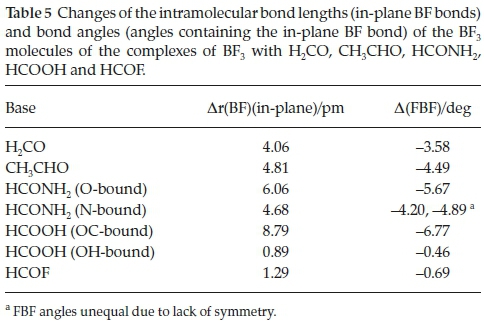
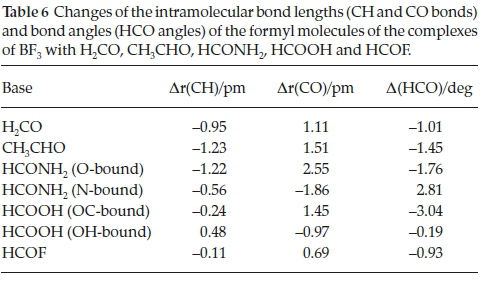
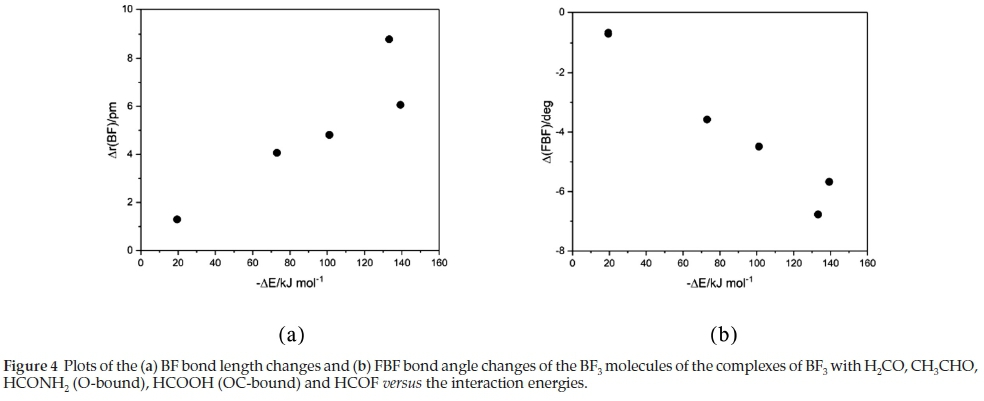
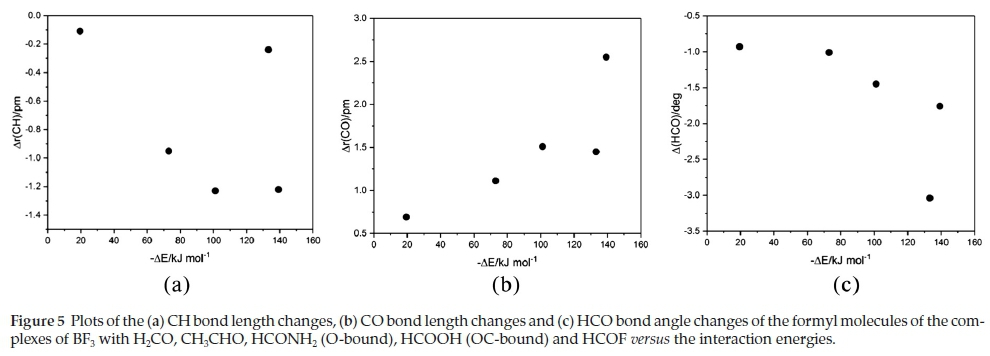
The changes undergone by the in-plane BF bonds and the FBF angles of the BF3 molecules on complexation are shown in Table 5. The BF bond lengths increase and the FBF angles decrease with strength of interaction; the FBF angle decreases are related to the degree of pyramidalization around the B atom. The intramolecular geometrical changes vary regularly with the interaction energies, as indicated in Fig. 4. Again, the outlier is the BF3.HCOOH complex, as a result of the unique bonding arrangement in this adduct. The corresponding intramolecular structural perturbations of the formyl bases (the CH and CO bond lengths and the HCO angles) are presented in Table 6. The H2CO base is a unique case, since the monomer possesses two equivalent CH bonds and HCO angles; the changes listed in Table 6 relate to the bond and angle involved in the interaction. The CH bonds contract, while the CO bonds extend and the HCO angles close up; as for the BF3 molecules, the intramolecular bond length and angle changes of the formyl molecules correlate in a regular manner with the interaction energies, as shown in Fig. 5. The BF3.HCOOH complex properties again fail to follow the trend.
3.3. Vibrational Spectra
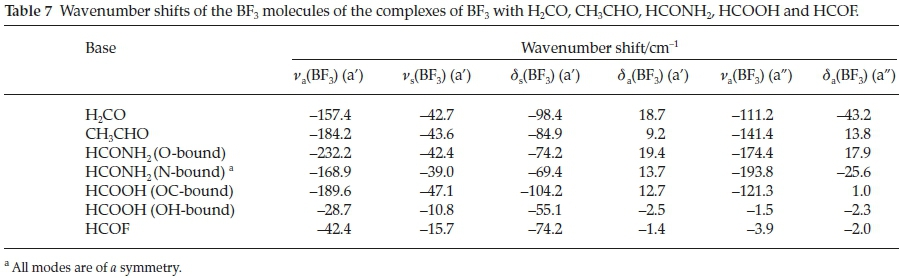
Table 7 reports the wavenumber shifts of the BF3 modes of the complexes. Only those of the antisymmetric stretching vibrations correlate sensibly with the interaction energies. Both the a' and a" modes undergo red shifts which increase with increasing strength of interaction. This relationship is illustrated in Fig. 6.
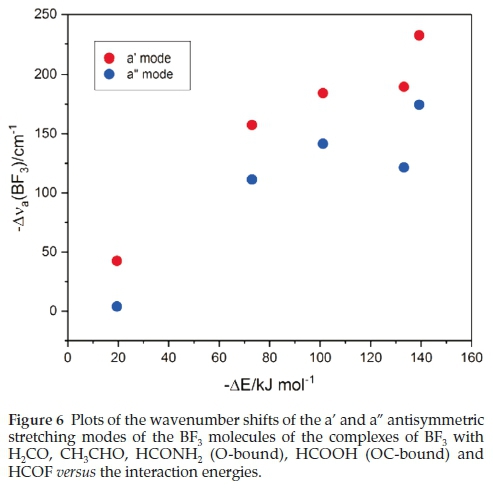
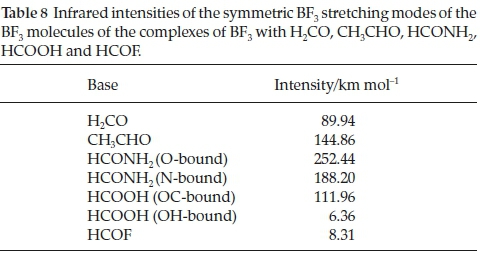
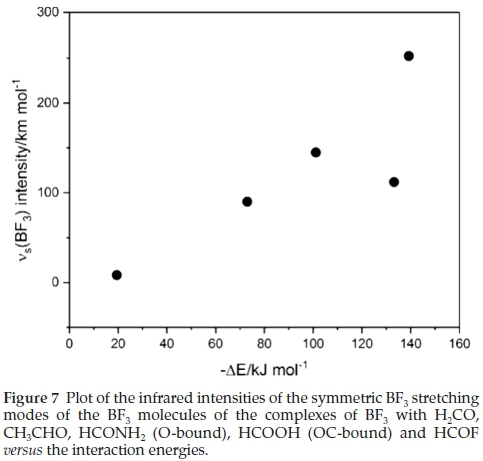
Associated with the wavenumber shifts of the BF3 modes are the perturbations of their infrared intensities. The complex/ monomer intensity ratios are rather insensitive to the interaction energies. The symmetric stretching mode, which is inactive in the monomer, gains intensity on complexation, as seen in Table 8, and the acquired intensity tracks with the strength of interaction, as evidenced by Fig. 7.
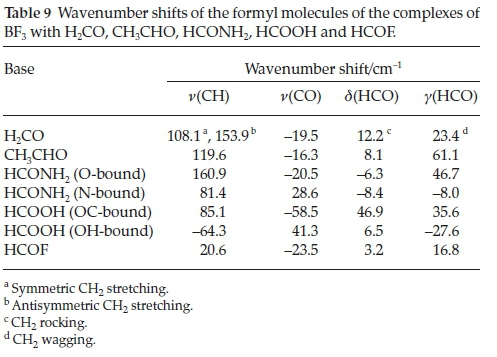
The corresponding data for the formyl moieties of the complexes, specifically the wavenumber shifts of the CH and CO stretching, the in-plane HCO bending (rocking) and the out-of-plane HCO bending (wagging) vibrations are collected in Table 9. These modes of the formyl molecules are the most characteristic vibrations of the bases and they would be expected to be influenced in a regular manner due to complexation. The CH modes are blue-shifted and the CO modes red-shifted, while the signs of the in-plane and out-of-plane bending mode shifts are inconsistent; there is no apparent dependence of the wave-number shifts on the nature of the base. This is largely due to the range of different structures exhibited by the base molecules. The intensity ratios of the formyl compound bases are also rather insensitive to the interaction energies. The perturbations of the vibrational spectra of the bases are therefore not reliable indicators of the extent of the interaction.
3.4. Natural Bond Orbital Analysis
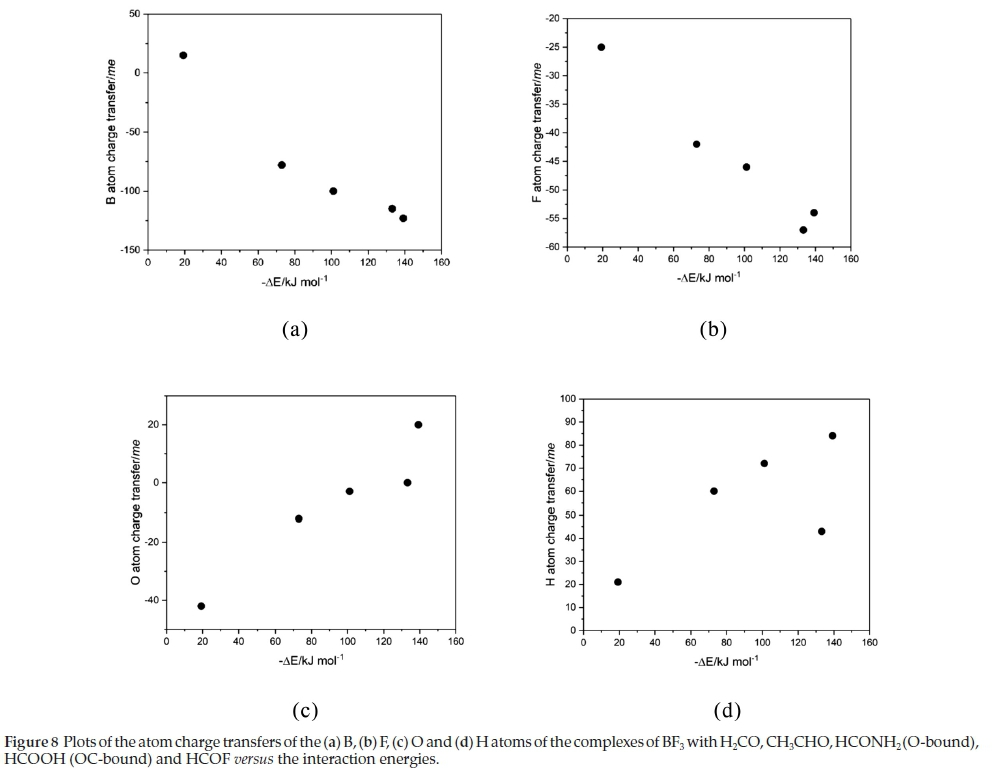
The NBO analysis revealed that the primary orbital interactions in each case were donation from an oxygen lone pair orbital to the π* orbital of BF3, and from a lone pair orbital of the in-plane fluorine atom to the o*(CH) orbital of the base. See Fig. 2 for an illustration of the modes of interaction for the various complexes. These primary interactions were accompanied by redistributions of charge within the five-membered rings such that the net changes are accumulation of charge for the B and F atoms and depletion for the O and H atoms. The charges for the B, F, O and H atoms directly engaged in the interactions are listed in Table 10, along with the transfers of charge per atom resulting from complex formation. The charge transfers for the B and F atoms become more negative, and those for the O and H atoms more positive, with increasing strength of interaction, as indicated in Fig. 8, confirming the internal consistency among the structures, vibrational spectroscopic changes, electronic charge redistributions and interaction energies in this family of complexes.
4. Results and Discussion (X = Li, BeH, BH2)
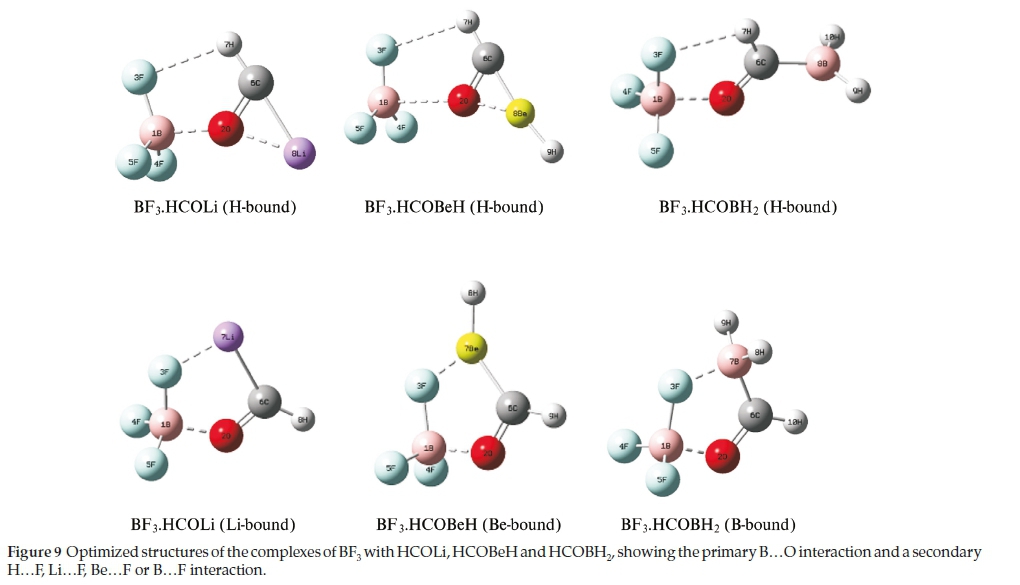
The optimized structures of the complexes of BF3 with the HCOLi, HCOBeH and HCOBH2 species are illustrated in Fig. 9. Two possible models were found for each complex. In addition to the primary interaction between the formyl oxygen and the boron atom, a secondary interaction was apparent, involving the in-plane fluorine atom and either the formyl hydrogen or the Li, Be or B atom. In addition, it should be noted that in the H-bound complexes with HCOLi and HCOBeH there is a weak internal bond between the O atom and the Li or Be atom, which is absent in the H-bound BF3.HCOBH2 adduct. This structural feature was also observed in the optimized structures of the HCOLi and HCOBeH monomers, apparent in Table 2. The Cartesian coordinates of the optimized structures are given in the Supplementary Information, in Tables S8 to S13.
4.1. Interaction Energies
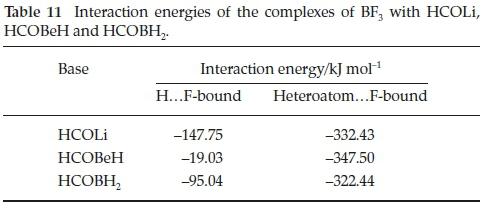
The interaction energies of the H...F-bound isomers are collected in Table 11. These energies are of the same order of magnitude as those of the complexes containing the H, CH3, NH2, OH and F atoms or groups. The energies, however, do not follow any trends related to the position of the heteroatom in the periodic table. The complexes with HCOLi and HCOBeH appear to belong to one group, characterized by the presence of the weak internal L1...O and Be...O bonds, while the complex with HCOBH2, in which this secondary interaction is absent, is more similar in properties to the adducts with H2CO, CH3CHO, HCONH2, HCOOH and HCOF (see Table 3). The interaction energies of the heteroatom.F-bound complexes are extremely large, and are virtually independent of the heteroatom.
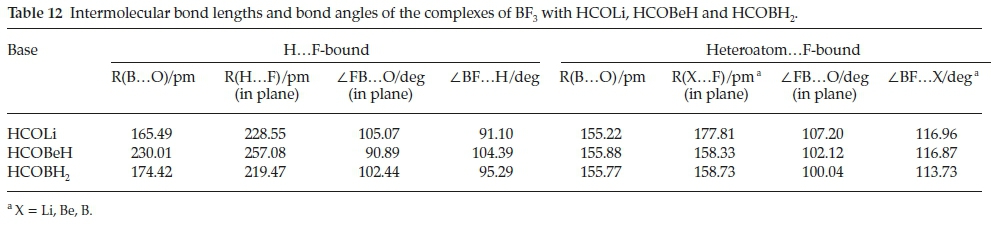
4.2. Molecular Structures
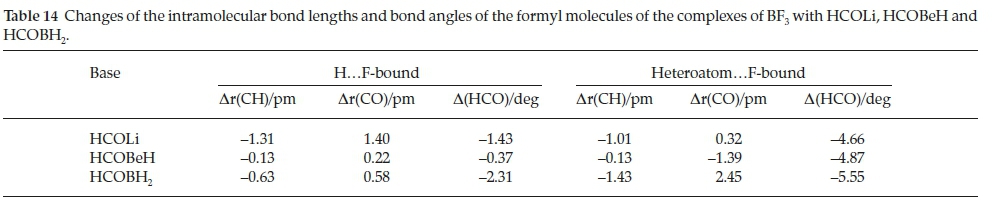
The intermolecular structural properties of this set of complexes are collected in Table 12. Consistent with the low value of the interaction energy of H.F-bound BF3.HCOBeH, the B.O distance is considerably longer than those of the Li and B analogues, coupled with the reversal of the magnitudes of the FB.O and BF.H angles, compared with BF3.HCOLi and BF3.HCOBH2. These properties have the effect of distorting the cyclic FBOCH arrangement, leading to a weakening of the interaction. The intermolecular parameters of the hetero-atom.F-bound adducts are remarkably constant, and reinforce the observation that the heteroatom.F interactions are significantly stronger than their H.F counterparts. The perturbations of the intramolecular bond lengths and bond angles relative to their monomer values are presented in Table 13. Confirming the contrast between the properties of the HCOBeH complex and those of the HCOLi and HCOBH2 analogues, the in-plane BF bond length and FBF angle are very similar to those in the monomer, while the extension of the bonds and the reduction of the angles for the other two bases are quite significant. The perturbations of the intramolecular geometrical properties of the second series of adducts are in line with the markedly larger interaction energies of this family. A similar pattern is found for the changes of the bond lengths and bond angles of the formyl molecules, with the H.F-bound bond length perturbations in the order HCOLi > HCOBH2 > HCOBeH (Table 14). As we found for the complexes with H2CO, CH3CHO, HCONH2, HCOOH and HCOF (Table 6), the CH bonds shorten and the CO bonds are extended, while the HCO angles contract. There is a much closer correspondence between the formyl molecule property changes of the H.F-bound and the heteroatom.F-bound groups than was the case for the BF3 molecules.
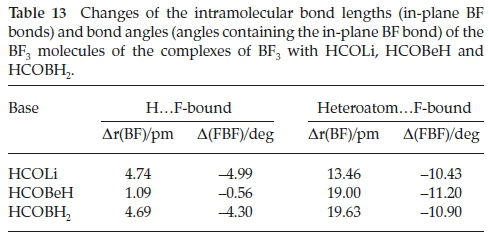
4.3. Vibrational Spectra
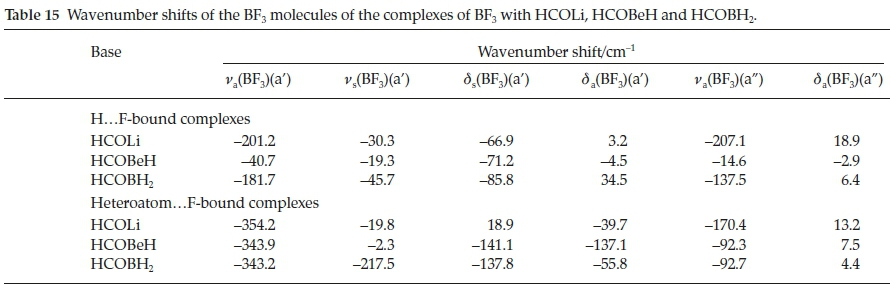
Table 15 presents the wavenumber shifts of the BF3 moieties of the HCOLi, HCOBeH and HCOBH2 complexes. For the H.F-bound series the three BF3 stretching modes are all red-shifted, with shifts in the sequence HCOLi > HCOBH2 > HCOBeH. This observation is in line with the order of the interaction energies. The bending modes behave inconsistently with respect to sign. The stretching vibrations of the hetero-atom.F-bound set are also red-shifted, as expected, but apart from the a' antisymmetric mode, where the shifts are remarkably similar, the order of magnitudes of the other two stretching modes is inconsistent. The same is true of the perturbations of the three bending mode wavenumbers. The induced infrared intensities of the symmetric BF3 stretching vibrations are listed in Table 16. Similar to the case of the complexes with H2CO, CH3CHO, HCONH2, HCOOH and HCOF (Table 8 and Fig. 7), the extent to which this mode is activated on complexation correlates with the interaction energy.
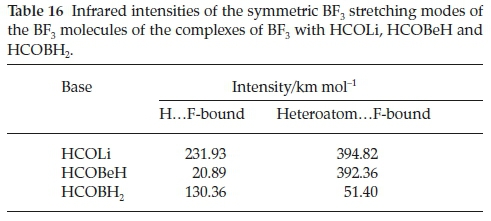
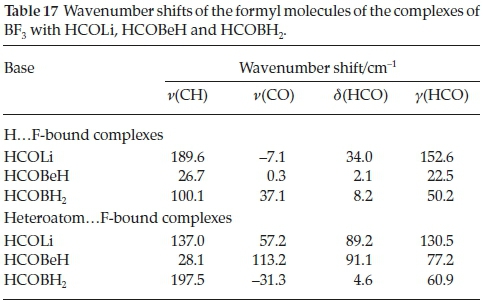
Table 17 shows the wavenumber shifts of the CH and CO stretching vibrations and of the in-plane and out-of-plane HCO bending modes. Among the H.F-bound complexes the CH stretching modes are blue-shifted, as was the case for the adducts with H2CO, CH3CHO, HCONH2, HCOOH and HCOF (Table 9), in the order HCOLi > HCOBH2 > HCOBeH, which correlates with the order of the interaction energies. The signs of the shifts of the CO stretching vibrations are variable. The two HCO bending modes, on the other hand, are uniformly blue-shifted, and the magnitudes of the shifts track with the interaction energies. The signs of the shifts of the hetero-atom.F-bound complexes are similar to those of the H.F-bound series, but their magnitudes do not correlate with the interaction energies. Consistent with the complexes with H2CO, CH3CHO, HCONH2, HCOOH and HCOF, the vibrational properties of the formyl moieties are not useful parameters for measuring the strength of interaction of these complexes.
4.4. Natural Bond Orbital Analysis
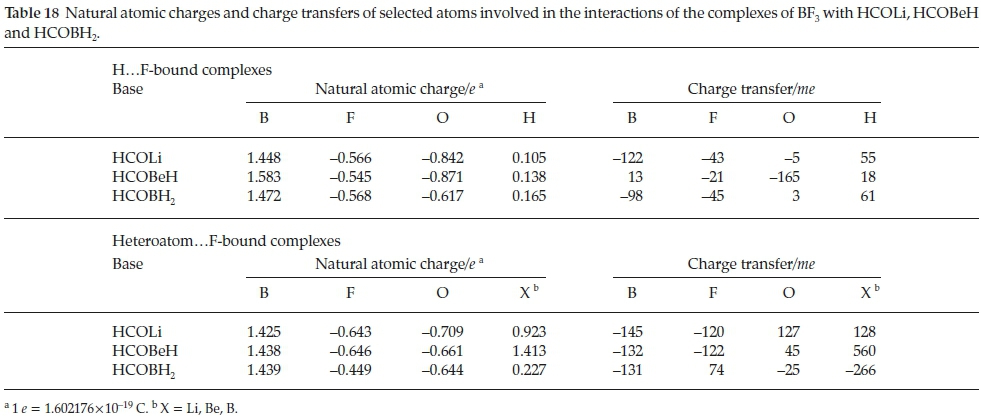
The natural atomic charges and their transfers are presented in Table 18. In the case of the H.. .F-bound species, the charge of the B atom in BF3.HCOBeH is more positive and that of the in-plane F atom less negative than those of BF3.HCOLi or BF3.HCOBH2. These features lead to a small positive charge transfer for the B atom and a very large negative change for the O atom. These results indicate a substantial charge equalization between the Be atom and the C and O atoms, which is sufficient to lower the interaction energy of BF3.HCOBeH considerably relative to the Li analogue. The extremely low interaction energy of BF3.HCOBeH, relative to BF3.HCOLi, may now be explained by the differences in the natural atomic charges of the Li and Be atoms, and their perturbations on complexation. The charges of the heteroatom.F-bound species are more regular than those of the H...F-bound analogues; it is in the values of the charge transfers that the contrast between the HCOLi, and HCOBeH complexes and that of the HCOBH2 adduct become apparent. While the charge transfers of the B atoms of BF3 are almost constant, there is a sign reversal for the F, O and X atoms between the HCOLi and HCOBeH species and the HCOBH2 complex. This contrast is due largely to the strong electrostatic interaction between the electropositive Li and Be atoms and the in-plane F atom, resulting from the partially ionic LiC and BeC bonds, while the B.F interaction is weaker, since the BC bond is predominantly covalent.
5. Conclusions
The properties (interaction energies, intermolecular geometries, intramolecular geometry changes and atomic charge trans fers) of both the BF3 and the HCOX components (X = H, CH3, NH2, OH and F) of the complexes correlate fairly well with one another. The exception is the BF3.HCOOH complex, whose structure features a secondary F.H interaction with the hydroxyl hydrogen atom, in contrast to the F.H interaction present in the other adducts, which is through the formyl hydrogen.
The wavenumber shifts of the antisymmetric stretching and the induced intensities of the symmetric stretching modes of the BF3 fragments track reasonably well with the complex interaction energies. The vibrational spectroscopic properties of the formyl moieties are not as well-behaved, and do not exhibit any dependence on the interaction energies.
The three complexes containing heteroatoms from groups 1,2 and 13 show two families of structures, one with a secondary H.F interaction and the other with a heteroatom.F linkage. The H.F-bound series have interaction energies comparable in magnitude with those of the complexes containing heteroatoms from groups 14 to 17. Those containing a heteroatom.. .F interaction, on the other hand, have much more substantial interaction energies, due to the partially ionic nature of the bonds involving the Li or Be atoms, and these energies are virtually independent of the nature of the heteroatoms.
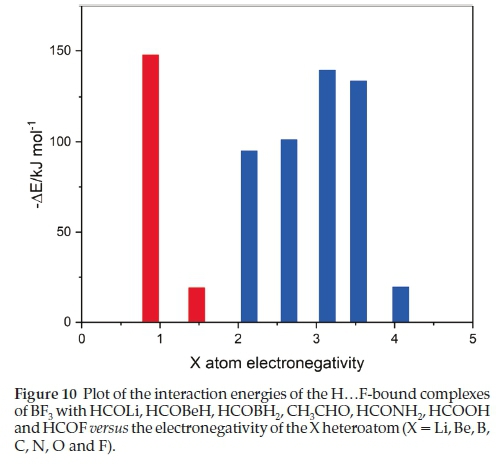
The H.F-bound adducts constitute a coherent set whose members possess interaction energies which vary as the heteroatom traverses the periodic table in an unpredictable way. These interaction energies are plotted against the Pauling electronegativities of the heteroatoms, exemplifying the group of the periodic table, in Fig. 10. The adducts fall into two groups, one containing heteroatoms from groups 1 and 2, and the other from groups 13 to 17. In the first series the interaction energy drops sharply from HCOLi to HCOBeH, for reasons related to the atomic charges, as discussed above. For the second series, the interaction energy rises from HCOBH2 to HCONH2, then falls off from HCONH2 to HCOF. These observations confirm that the physical properties of related series of molecules or complexes are usually governed by a complicated interplay of properties of the members of the series, such as the gas phase basicities, the mean polarizabilities and the group electronegativities of the X moieties, for example, and these features can balance one another in an unexpected fashion, with the HCOBH2,CH3CHO and HCONH2 set determined mainly by one particular property and the HCONH2, HCOOH and HCOF family by another. The natures of these interacting properties are a matter for speculation.
Supplementary Material
Supplementary information is provided in the online supplement.
Acknowledgements
This work is based upon research supported in part by the National Research Foundation of South Africa (NRF) under Grant Number 2053648. The grantholder acknowledges that any opinions, findings and conclusions or recommendations expressed in any publication generated by NRF-supported research are those of the author and that the NRF accepts no liability in this regard. The author also acknowledges the University of KwaZulu-Natal for financial assistance and the Centre for High Performance Computing (Cape Town) and the University of KwaZulu-Natal Hippo computer cluster for the use of computational facilities.
References
1 D.R. Martin, Coordination compounds of boron trichloride. I. A review, Chem. Rev., 1944, 34, 461-473. [ Links ]
2 D.R. Martin, Coordination compounds of boron bromide and boron iodide, Chem. Rev, 1948, 42, 581-599. [ Links ]
3 N.N. Greenwood and R.L. Martin, Boron trifluoride coordination compounds. Quart. Rev. Chem. Soc., 1954, 8, 1-39. [ Links ]
4 P. Hobza and K. Müller-Dethlefs, Non-covalent interactions. Theory and experiment, RSC Publishing, Royal Society of Chemistry, Cambridge, 2010. [ Links ]
5 J.S. Murray, P. Lane, T. Clark, K.E. Riley and P. Politzer, ó-Holes, p-holes and electrostatically-driven interactions, J. Mol. Model., 2012, 18, 541-548. [ Links ]
6 S. Grabowski, Triel bonds, p-hole-p-electrons interactions in complexes of boron and aluminium trihalides and trihydrides with acetylene and ethylene, Molecules, 2015, 20, 11297-11316. [ Links ]
7 W.M. Latimer and WH. Rodebush, Polarization and ionization from the standpoint of the Lewis theory of valence, J. Am. Chem. Soc., 1920, 42, 1419-1433. [ Links ]
8 D.N. Shigorin, Infrared absorption spectra study of H-bonding and of metal-element bonding, Spectrochim. Acta, 1959,14, 198-212. [ Links ]
9 J.-M. Dumas, H. Peurichard and M.J. Gomel, CX4.. .base interactions as models of weak charge transfer interactions - comparison with strong charge transfer and hydrogen-bond interactions, J. Chem. Res. -S, 1978, 54-57. [ Links ]
10 W. Wang, B. Ji and Y. Zhang, Chalcogen bond: a sister noncovalent bond to halogen bond, J. Phys. Chem. A, 2009,113, 8132-8135. [ Links ]
11 M. Yanez, P. Sanz, O. Mo, I. Alkorta and J. Elguero, Beryllium bonds, do they exist?, J. Chem. Theory Comput, 2009, 5, 2763-2771. [ Links ]
12 S. Zahn, R. Frank, E. Hay-Hawkins and B. Kirchner, Pnicogen bonds: a new molecular linker?, Chem. - Eur. J, 2011, 17, 6034-6038. [ Links ]
13 A. Bauza, T.D. Mooibroek and A. Frontera, Tetrel-bonding interaction: rediscovered supramolecular force?, Angew, Chem. Intern. Edit. Engl., 2013, 52, 12317-12321. [ Links ]
14 T. Clark, M. Hennemann, J.S. Murray and P. Politzer, Halogen bonding: the ó-hole, J. Mol. Model., 2007,13, 291-296. [ Links ]
15 T.A. Ford, The vibrational spectra of the boron halides and their molecular complexes. Part 13. Ab initio studies of the complexes of boron trifluoride with formaldehyde and some of its analogs, Int. J. Quantum Chem, 2012,112, 478-488. [ Links ]
16 M.J. Frisch, G.W Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski and D.J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2009. [ Links ]
17 C. M0ller and M. S. Plesset, Notes on an approximation treatment for many-electron systems, Phys. Rev., 1934, 46, 618-622. [ Links ]
18 R.A. Kendall, T.H. Dunning, Jr. and R.J. Harrison, Electron affinities of the first row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992,96, 6796-6806. [ Links ]
19 B. Liu and A.D. McLean, Accurate calculations of the attractive interactions of two ground state beryllium atoms, J. Chem. Phys., 1973, 59, 4557-4558. [ Links ]
20 S.F. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970,19, 553-556. [ Links ]
21 E.D. Glendening, J.K. Badenhoop, A.E. Reed, J.E. Carpenter, J.A. Bohmann, C.M. Morales and F. Weinhold, NBO Version 3.1, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2009. http://www.chem.wisc.edu/~nbo5 (accessed 4 August 2010). [ Links ]
Received 20 April 2018
Revised 3 September 2018
Accepted 10 October 2018
* To whom correspondence should be addressed. E-mail: ford@ukzn.ac.za
Dedicated to the memory of Daniel Thomas Hindler, 19 May 1989 - 27 October 2017
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}