










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.69 Durban 2016
http://dx.doi.org/10.17159/0379-4350/2016/v69a9
RESEARCH ARTICLE
Organocatalyzed Mannich Reactions on Minocycline: Towards Novel Tetracycline Antibiotics
Tirivashe E. ChiwunzeI; Rafiatu AzumahI; Melissa RamtahalI; Anou M. SomboroI; Per I. ArvidssonII; Hendrik G. KrugerI; Thavendran GovenderI, *; Tricia NaickerI, *
ISchool of Pharmacy and Pharmacology, University of KwaZulu-Natal, Westville Campus, Durban 4000, South Africa
IIScience for Life Laboratory, Drug Discovery and Development Platform, and Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden
ABSTRACT
Herein, we report the development of a mild synthetic route towards novel minocycline derivatives using the proline-catalyzed three-component Mannich reaction. The reaction conditions were optimized and was then screened for its tolerance to the other popular organocatalysts as well as variation of the ketone and aldehyde substrates. The Mannich adducts were evaluated for their in vitro antibacterial efficacies against Gram-negative and Gram-positive bacteria.
Keywords: Minocycline,tetracycline, antibiotics, Mannich reaction, organocatalysis
1. Introduction
The first class of tetracyclines was introduced in the 1950s as safe and effective broad-spectrum antibiotics. These agents have been important medical products for the last 50 years;1 however, the misuse of antibiotics has accelerated the natural inevitable phenomenon of resistant strain evolution. Tetracyclines inhibit bacterial growth by interfering with protein synthesis. They attach themselves to the bacterial 30S subunit of the rRNA (A site) which results in the prevention of the binding of amino-acyl-tRNA.2 The emergence of resistant bacteria to many of the current antibiotics is a major worldwide threat to public health.
There is an urgent need for the identification and development of novel antibiotics with new modes of action and significant therapeutic efficacies against these pathogens.3 Modification of existing antibiotic classes to new derivatives is an option to overcome this rapid drug resistance development. The urgent need to revive the tetracycline has brought about renewed interest to this class of compounds.
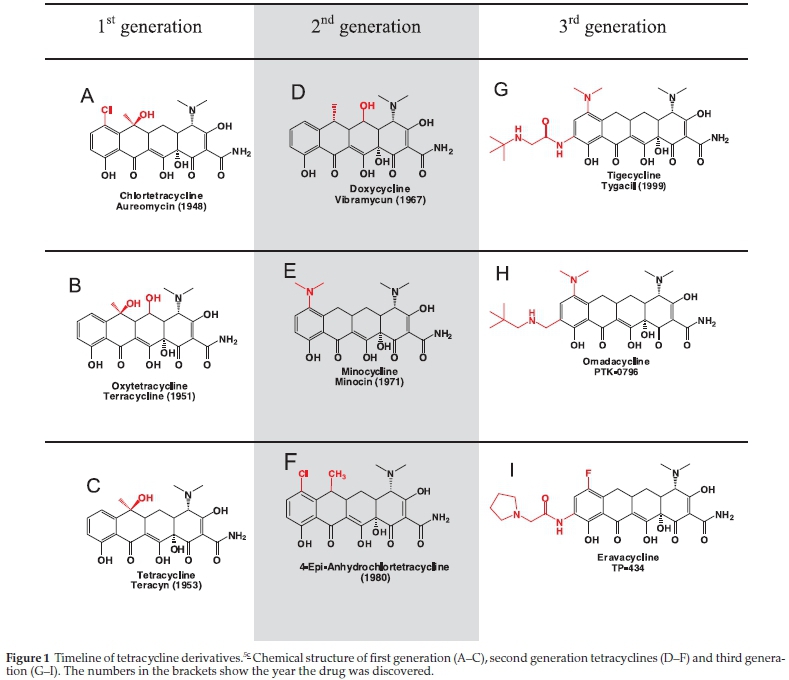
Most of the early tetracyclines were naturally occurring molecules (first generation), e.g. from Streptomyces aureofaciens and Streptomyces rimosus, or a product of semi-sythethic methods (second generation), e.g. minocycline and doxycycline (Fig. 1). Despite the great success of the early tetracyclines, improvements had to be made to better the pharmacokinetic properties, antimicrobial potency and also to decrease toxicity. This prompted the development of the third generation,which has led to many more derivatives and to the discovery of tigecycline, used in the fight against resistant strains.4 Although tigecycline is the first tetracycline drug to be brought to the market in the last half century, two more tetracyclines (omadacycline and eravacycline) are following in phase III clinical trials.5
Structure-activity relationship (SAR) studies6 on the tetracycline backbone have shown that it can tolerate structural modifications on positions C4 to C9 (Fig. 1).7 These chemical modifications often result in the change of chemical-physical properties and improved ability to overcome drug efflux. Recently K.C. Nicolaou et al. (2014), CuixiangSun et al. (2015) and Hrvoje Petkovic etal. (2015) reported novel tetracycline derivatives with extensions on the C and D rings.8 These compounds showed antibacterial activity against a wide range of clinically important bacterial isolates, including multidrug-resistant, Gram-negative pathogens.8a,8b
Given the recent need and synthetic reports on the tetracycline backbone to yield more potent derivatives such as tigecycline and omadacycline, we envisioned an easy entry to new derivatives based on the commercially available minocycline scaffold through milder synthetic routes.
Biocatalysts and metal complexes are amongst the most popular ways to synthesize optically pure molecules. Asymmetric organocatalysis was developed, in the past decade, as the need for alternatives that are less costly, use of less toxic reagents and more environmental friendly conditions.9 The last few years have witnessed a spectacular advancement in this field for the synthesis of medicinally important chiral.9 A substantial number of reactions using organocatalysis have been developed that were equivalent or better than conventional synthetic methods. Amongst the approaches that benefited are the well-known Aldol10, Mannich11 and Michael12 reactions, which are powerful strategies in synthetic organic chemistry, as they allow the formation of new C-C bonds.13 Recently there is a steady increase in the number of reports that are applying organocatalysis in the field of medicinal chemistry due to its mild and relatively benign approach to asymmetric synthesis.14
We have recently reported on the application of organo-catalysis in the synthesis of novel β-lactam derivatives.13b15 Herein we report the first organocatalyzed Mannich reactions, to make the D-ring substituent modification, on the minocycline scaffold to increase substituent bulk that can mimic the steric hindrance achieved by tigecycline. The end products of the Mannich reactions are also known as beta-amino ketones.16 These compounds have shown the ability to increase the hydro-philic properties of drugs through the introduction of a polar function in their structure.17 In other studies, they have been shown to act as prodrugs, releasing the active substance under controlled hydrolytic conditions via deaminomethylation18 or deamination.19 In addition to the organocatalyzed synthesis of new minocycline derivatives, preliminary biological evaluation of these compounds is also outlined.
2. Results and Discussion
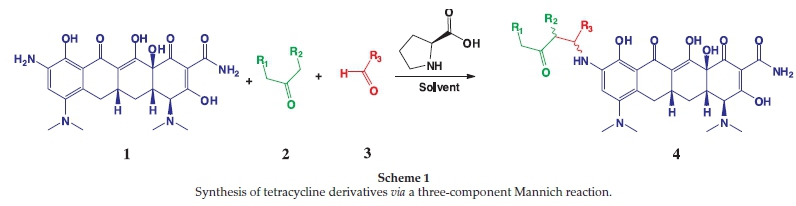
We successfully carried out a proline catalyzed three component Mannich reaction on the commercially available 9-amino minocycline core witrious aldehydes and ketones (Scheme 1).
The reaction was optimized using a 9-amino minocycline (1), hydroxyl acetone (2) and nitro-benzaldehyde (3) in a one-pot reaction (Table 1). Proline was initially chosen to optimize the reaction as it has been well established to catalyze similar trans-formations.13a,20 Solvent screening began with dimethylsulphoxide (DMSO) since it is the most commonly reported solvent for Mannich reactions. However, in this case it gave poor conversion (41 %), which was probably due to the partial solubility of 1. Other polar solvents such as ethanol, methanol, 2-isopropanol and dimethylformamide (DMF) in which the amine 1 was more soluble was attempted. DMF gave poor conversion (57 %) despite being able to fully solubilize 1. The alcoholic solvents exhibited much improved results in the range of 76-96 %, with methanol as the best solvent (96 % conversion) under room temperature conditions and 8 hours of reaction time.
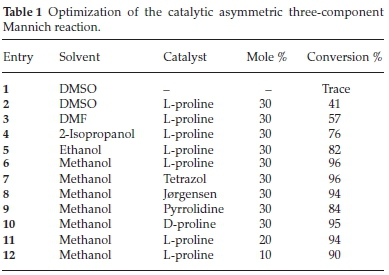
In order to examine further catalyst effects, we selected a few popular amine catalysts (proline, tetrazol, pyrrolidine and Jorgensen catalyst) that are known to facilitate the progression of Mannich reactions21. All of the catalysts showed good activities with conversions in the range of 84-96 %; in alignment with past reports proline typically gave superior results.11a,20,22 The amount of proline could also be effectively reduced to 10 mol % while still obtaining good product conversions (>90 %) in a reasonable reaction time (<24 h).
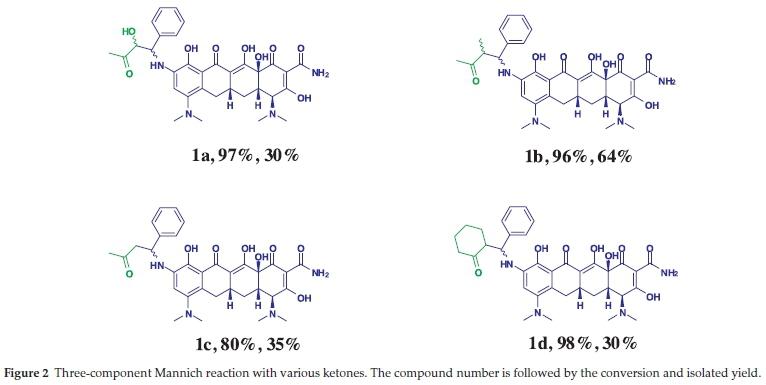
As previously shown by B. List el al. (2001)6a, that a variety of ketones can be used in such proline-catalyzed Mannich reactions. Reacting four different ketones (butanone, methoxyacetone, ethyl methyl ketone, acetone and hydroxyacetone) with 9-amino minocycline and benzaldehyde furnished the desired products with high conversions 80-98 % (Fig. 2). The aliphatic (1a-1c) and cyclic ketones (1d) generated the desired products while aromatic ketones only produced trace amounts on the HPLC-MS.
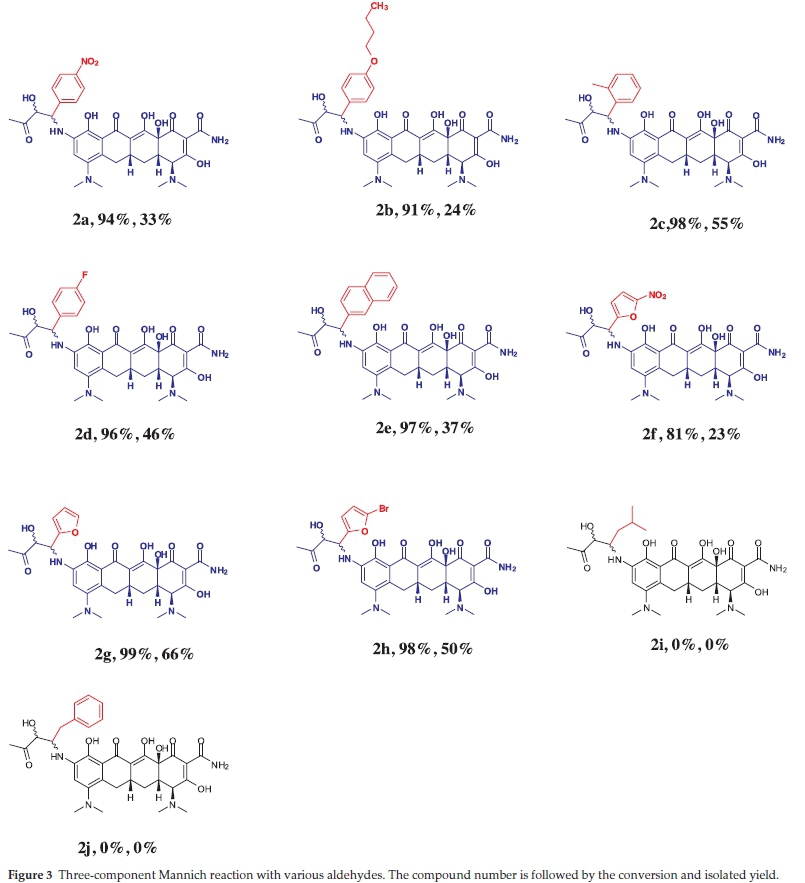
We next performed an aldehyde screen for the three-component Mannich reactions (Fig. 3). Among the aldehydes screened the α-branched aromatic aldehydes (2a-2h) provided the Mannich products in reasonable yields. Not only benzaldehyde derivatives but also heteroaromatic aldehydes (2f, 2g and 2h) worked well. Typically α-branched aldehydes are more efficient substrates than the α-unbranched aldehydes11a,23 but in our case; α-unbranched aldehydes (2i and 2j) only afforded trace amounts of the corresponding Mannich adducts.
The proton and especially the 13C NMR data of the starting material and related products appeared to be problematic. Upon a literature search it was found that the majority of reports did not provide 13C spectral data.8b24 It appears that specialized automated triple broadband (ATB) probes are required for the generation of quality 13C spectra.8c,25
Similarly, the proton spectra were broad and it was difficult to deduce much about the diastereomeric details of the products. Based on the proton spectra as well as HPLC results, it appears that the products may be formed largely as one diastereomer.
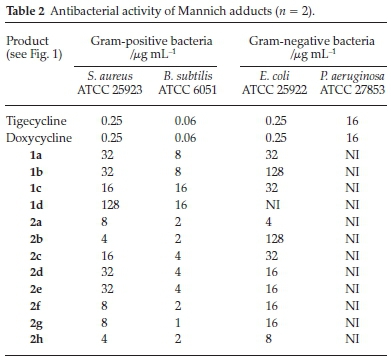
The activities of the above new tetracycline derivatives were determined by the agar dilution method following the recommendations of the National Committee for Clinical Laboratory Standards26. The analogues tested exhibited antibacterial efficacies against both Gram-positive and Gram-negative bacteria (Table 2).
2.1. Antibacterial Activity
Tigecycline and Doxycycline were used as control drugs and their MIC values were in agreement with the Clinical Laboratory Standards Institute (CLSI).40 The Mannich adducts in general showed good activities against Gram-positive bacteria, with the best giving a MIC of 1.0 mL-1 against B. sublilis. However, these compounds were always less active against Gram-negative bacteria; none of them showed activity against P. aeruginosa and the MICs were >16 mL-1 against E.coli with only a couple of exceptions. The poor activities of these compounds against these strains could be as a result of low permeationof the Gram-negative cell wall.
Compounds 1a, 1b, 1c and 1d showed the effect of changing the ketone on the adducts, with the most polar exhibitiing the better activity. The aldehydes were then varied and the benzaldehyde derivatives demonstrated a mentionable structural-activity interrelation. Addition of a substitution group onto the benzene ring proved to be crucial for the antibacterial activity. Compounds 2a, 2b and 2d have electron-withdrawing substituents on the para position of the benzene ring and exhibited significant improvements compared to the non-substituted 1a. Compound 2b exhibited an enormous drop in potency for the Gram-negative bacteria. This could be due to the hydro-phobicity of the extended carbon chain, which makes it difficult for the compound to pass through the peptidoglycan layer of the cell due to increased hydrophobic interactions. Increasing the bulkiness of the benzene ring had a less significant effect on the potency of this compound as seen by compounds 2c(methyl group) and 2e(fused benzene ring). The furfuraldehyde derivatives gave poor structural-activity correlation, as the addition of a substitution group to the furfural ring (2f, 2g and 2h) did not have much influence on the MICs.
3. Conclusions
In summary, we have shown for the first time that organo-catalysis can be employed to synthesize tetracycline derivatives. Although, comparsion to the parent minocycline showed lack of benefit from the addition of the alkylamino side chains at the C9 position, this methodology shows a new and mild synthetic route to derivatize this family of compounds and could be a starting point for future analogues with better biological activity. The Mannich adducts were more active against Gram-positive bacteria than they are with Gram-negatives. The structure-activity relationship studies of these compounds provided useful information on the structural requirements for activity against Gram-negative bacteria. It also indicated that it is possible to design compounds with selectivity just against Gram-positive bacteria. The promising in vilro activity of some of the compounds (e.g. 2a and 2g) makes them potential candidates for the development of new antibiotics selectively targeting Gram-positive pathogens.
4. Experimental Section
4.1. General Experimental Procedures
Reagents and solvents were purchased from Sigma-Aldrich and were used without purification unless otherwise stated. High resolution mass spectrometric data were obtained with a Bruker micrOTOF-Q II instrument that operated at ambient temperatures and at a sample concentration of ca. 1.0 ppm. NMR spectra were recorded with Bruker AVANCE III400 or 600 MHz instruments at room temperature. Chemical shifts are expressed ppm, the deuterated solvent reference peak were used as an internal standard for autocalibration.
4.2. General Experimental Procedure for the Synthesis of Mannich Products from Hydroxyacetone
To a suspension of L-proline (4.2 mg, 0.3 mmol) in methanol (2.0 mL), 9-amino minocycline hydrochloride (60 mg, 1.1 mmol) was added. To this mixture, the aldehyde (1.0 mmol) followed by the ketone (1.0 mL) was added and stirred at room temperature for overnight. The reactions were then directly purified straight using semi-preparative reverse-phase HPLC.
4.3. Chromatography Method
Semi-preparative reverse-phase HPLC (Shimadzu, Japan) was conducted usingaACE C18 preparative column (150 x 21.2mm) using gradient of0%Bfor5min, 0%Bto85%Bover 55 min, 85%Bto95%B over 1 min, 95 % B for 5 min, 95%Bto0%B over 1 min, 0%Bfor5min (A = dH2O with 0.1 % FA; B = Methanol with 0.1 % FA; flow rate =15 mL min-1;A254 nm). The fractions were characterized by LCMS (Shimadzu, Japan). Fractions which showed desired mass were collected and concentrated under reduced pressure and lyophilized to give the products as dark red powders. The products are hygroscopic and also decompose upon heating, therefor melting points were not obtained.
4.4. Antibacterial Assays
MICs of the tested compounds were determined by broth microdilution, according to CLSI guidelines. Briefly, two fold dilutions of each inhibitor was made in cation adjusted Mueller-Hinton Broth (CAMHB) in a microtitre plate. A 10 mL of 0.5 McFarland bacteria inoculum was added to make a total volume of 100ml in each microtitre well. Plates were then incubated at 37 ° for 18-22 h under aerobic conditions. MICs were defined at the lowest concentration of antimicrobial agent inhibiting visible growth using the Alamar blue assay. S. aureus ATCC 25923, B. subtilis ATCC 6051, E. coli ATCC 25922 and P. aeruginosa ATCC 27853 were used in this study. Control wells of the amount of DMSO used were also done. Assays were done in duplicate to confirm results.26
4.5. NMR Data
OH and NH protons did not show on the NMR spectra.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-9-(2-hydroxy-3-oxo-1-phenylbutylamino)-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (1a)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.45-7.36 (m, 2H), 7.33-7.28 (m, 1H), 7.28-7.23 (m, 1H), 7.23-7.18 (m, 1H), 6.88-6.75 (m, 1H), 4.89-44.74 (m, 1H), 4.52^.21 (m, 1H), 3.23-2.82 (m, 14H), 2.37-2.21 (m, 3H), 2.21-2.13 (m, 1H), 2.08-1.91 (m, 2H), 1.66-147 (m, 1H), 1.32-1.24 (m, 1H), (OH's, 5, NH's, 3) . HRMS (ESI+): calcd. for C33H39N4O9 [M + H]+ 635.2712; found 635.2724.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-9-(2-methyl-3-oxo-1-phenylbutylamino)-1,11-dioxo-1 ,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (1b)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.45-7.35 (m, 2H), 7.34-7.26 (m, 2H), 7.24-7.18 (m, 1H), 6.75-6.68 (m, 1H), 4.85-4.78 (m, 1H), 4.11-3.97 (m, 1H), 3.17-2.80 (m, 17H), 2.52-2.23 (m, 2H), 2.20-2.16 (m, 1H), 2.00-1.96 (m, 1H), 1.64-1.48 (m, 1H), 1.28-1.22 (m, 1H), 1.08-1.00 (m, 1H), 1.00-0.906 (m, 1H), (OH's, 4, NH's, 3).
HRMS (ESI+): calcd. for C34H41N4O8 [M+H]+ 633.2919; found 633.2906.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-1,11-dioxo-9-(3-oxo-1-phenylbutylamino)-1,4,4a,5,5a, 6,11,12a-octahydrotetracene-2-carboxamide (1c)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.63-7.59 (m, 2H), 7.41-7.38 (m, 2H), 7.33-7.27 (m, 1H), 6.77-6.70 (m, 1H), 4.867-4.75 (m, 1H), 3.25-3.19 (m, 8H), 3.08-2.97 (m, 12H), 2.48-2.38 (m, 1H), 2.37-2.32 (m, 1H), 2.19-2.10 (m, 2H), (OH's, 4, NH's, 3). HRMS (ESI+):calcd. for C33H39N4O8 [M + H]+ 619.2762; found 6.19.2751.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-1,11-dioxo-9-((1R)-(2-oxocyclohexyl)(phenyl)methyl-amino)-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (1d)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.47-7.40 (m, 2H), 7.36-7.26 (m, 2H), 7.24-7.17 (m, 1H), 6.82-6.75 (m, 1H), 4.90-4.78 (m, 1H), 4.12-4.04 (m, 1H), 3.11-2.96 (m, 14H), 2.36-2.28 (m, 4H), 1.73-1.56 (m, 9H), (OH's, 4, NH's, 3). HRMS (ESI+): calcd. for C36H43N4O8 [M + H]+ 659.3362; found 659.3352.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-9-((1S,2R)-2-hydroxy-1-(4-nitrophenyl)-3-oxobutyl-amino)-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (2a)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 8.20-8.15 (m, 2H), 7.72-7.66 (m, 2H), 6.94-6.88 (m, 1H), 4.49-4.45 (m, 1H), 4.1L4.03 (m, 1H), 3.13-2.98 (m, 14H), 2.37-2.29 (m, 3H), 2.14-2.08 (m, 2H), 1.64-1.53 (m, 1H), 1.33-1.27 (m, 2H), (OH's, 5, NH's, 3). HRMS (ESI+): calcd. for C33H38N5O11 [M + H]+ 680.2562; found680.2554.
(4S,4aS,5aR,12aS)-9-((1S,2R)-1-(4-butoxyphenyl)-2-hydroxy-3-oxobutylamino)-4,7 bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (2b)
Ή NMR (600 MHz, CD3OD + 0.1 % TFA) 7.36-7.28 (m, 2H), 6.87-6.83 (m, 2H), 6.82-6.77 (m, 1H), 4.43^.38 (m, 1H), 4.11-4.04 (m, 1H), 3.93-3.86 (m, 2H), 3.15-2.96 (m, 15H), 2.30-2.23 (m, 3H), 2.21-2.15(m, 1H), 2.08-2.03 (m, 1H), 1.71-1.65 (m, 2H), 1.63-1.56 (m, 1H), 1.47-1.41 (m, 2H), 1.39-1.36 (m, 1H), 0.95-0.90 (m, 3H), (oH's, 5, NH's, 3). HRMS (ESI+): calcd. for C37H47N4O10 [M + H]+ 707.3287; found 707.3270.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-9-((1S,2R)-2-hydroxy-3-oxo-1-o-tolylbutylamino)-1,1 1-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (2c)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.23-7.17 (m, 2H), 7.14-7.11 (m, 1H), 7.09-7.05 (m, 1H), 6.51-6.48 (m, 1H), 4.90-4.74 (m, 1H), 4.392^.35 (m, 1H), 3.05-2.96 (m, 15H), 2.59-2.56 (m, 3H), 2.51-2.47 (m, 1H), 2.35-2.32 (m, 3H), 1.99-1.97 (m, 1H), 1.63-1.53 (m, 2H), (OH's, 5, NH's, 3). HRMS (ESI+): calcd. for C34H41N4O9 [M + H]+ 649.2868; found 649.2866.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-9-((1S,2R)-1-(4-fluorophenyl)-2-hydroxy-3-oxobutylamino)-3,10,12,12a-tetra-hydroxy-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (2d)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.52-7.41 (m, 2H), 7.10-6.97 (m, 2H), 6.91-6.81 (m, 1H), 4.47-4.37 (m, 1H), 4.18-4.01 (m, 1H), 3.21-2.92 (m, 17H), 2.35-2.29 (m, 3H), 2.10-2.06 (m, 1H), 1.68-1.55 (m, 1H), (OH's, 5, NH's, 3). HRMS (ESI+): calcd. for C33H38FN4O9 [M + H]+ 653.2617; found 653.2592.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-9-((1S,2R)-2-hydroxy-1-(naphthalen-2-yl)-3-oxobutyl amino)-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (2e)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.93-7.90 (m, 1H), 7.80-7.78 (m, 2H), 7.61-7.58 (m, 1H), 7.45-7.40 (m, 3H), 6.91-6.88 (m, 1H), 4.85-4.79 (m, 1H), 4.56^.52 (m, 1H), 3.04-2.95 (m, 14H), 2.35-2.32 (m, 3H), 2.25-2.22 (m, 1H), 2.19-2.16 (m, 1H), 2.078-2.036 (m, 1H), 2.02-2.00 (m, 1H), 1.59-1.53 (m, 1H), (OH's, 5, NH's, 3) . HRMS (ESI+): calcd. for C37H41N4O9 [M + H]+685.2868; found 685.2865.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-3,10,12,12a-tetra-hydroxy-9-((1R,2R)-2-hydroxy-1-(5-nitrofuran-2-yl)-3-oxo-butylamino)-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetra-cene-2-carboxamide (2f)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.41-7.27 (m, 2H), 6.73-6.65 (m, 1H), 4.90-4.80 (m, 1H), 4.74^.65 (m, 1H), 3.28-3.18 (m, 10H), 3.08-3.00 (m, 7H), 2.34-2.30 (m, 3H), 1.72-1.58 (m, 2H), (OH's, 5, NH's, 3). HRMS (ESI+): calcd. for C31H36N5O12 [M + H]+ 670.2355; found 670.2322.
(4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-9-((1R,2R)-1-(fura-2 -yl)-2-hydroxy-3-oxobutylamino)-3,10,12,12a-tetrahydroxy-1, 11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carbox-amide (2g)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.51-7.35 (m, 1H), 7.17-7.05 (m, 1H), 6.38-6.23 (m, 2H), 4.86^.80 (m, 1H), 4.65-4.56 (m, 1H), 3.22-3.15 (m, 10H), 3.09-2.98 (m, 7H), 2.41-2.32 (m, 1H), 2.30-2.24 (m, 2H), 2.22-2.19 (m, 1H), 1.63-1.57 (m, 1H), (OH's, 4, NH's,3). HRMS (ESI+): calcd. for C31H37N4O10 [M + H]+ 625.2504; found 625.2486.
(4S,4aS,5aR,12aS)-9-(1-(5-bromofuran-2-yl)-2-hydroxy-3-oxo-butylamino)-4,7-bis (dimethylamino)-3,10,12,12a-tetra-hydroxy-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide (2h)
1H NMR (600 MHz, CD3OD + 0.1 % TFA) 7.22-7.08 (m, 1H), 6.39-6.20 (m, 2H), 4.87-4.78 (m, 1H), 4.61^.52 (m, 1H), 3.27-3.11 (m, 10H) 3.09-2.92 (m, 7H), 2.44-2.33 (m, 1H), 2.30-2.21 (m, 3H), 1.67-1.54 (m, 1H), (OH's, 5, NH's, 3). HRMS (ESI+): calcd. for C31H36Br N4O10 [M + H]+ 703.1609; found 703.1588.
Acknowledgements
This study was supported by College of Health Science, University of Kwa-Zulu Natal, Durban, South Africa; Aspen Pharma-care, South Africa, and the South African National Research Foundation.
Supplementary Material
For copies of proton NMR spectra, HPLC purity chromato-graphs and high-resolution mass spectra.
References
1 B.M. Duggar, Aureomycin: a product of the continuing search for new antibiotics, Ann. N.Y. Acad. Sci., 1948, 51, 177-181. [ Links ]
2 a) I. Chopra, P. Hawkey and M. Hinton, Tetracyclines, molecular and clinical aspects, J. Antimicrob. Chemother., 1992, 29, 245-277; [ Links ] b) D. Schnappinger, W. Hillen, Tetracyclines: antibiotic action, uptake, and resistance mechanisms, Arch. Microbiol., 1996, 165, 359-369. [ Links ]
3 A. Parisien, B. Allain, J. Zhang, R. Mandeville and C. Lan, Novel alternatives to antibiotics: bacteriophages, bacterial cell wall hydrolases, and antimicrobial peptides, J. Appl. Microbiol., 2008,104, 1-13. [ Links ]
4 P.J. Petersen, N. Jacobus, W Weiss, P. Sum and R. Testa, In vitro and in vivo antibacterial activities of a novel glycylcycline, the 9-t-butylgly-cylamido derivative of minocycline (GAR-936), Antimicrob. Agents Chemother., 1999,43, 738-744. [ Links ]
5 a) M.G. Charest, D. R. Siegel and A.G. Myers, Synthesis of (-)-tetracycline, J. Am. Chem. Soc., 2005,127,8292-8293; [ Links ] b) Y. Wang, R. Castaner, J. Bolos and C. Estivill, Amadacycline tetracycline antibiotic, Drugs of the Future, 2009, 34, 11-15; [ Links ] c) F. Nguyen, A.L. Starosta, S. Arenz, D. Sohmen, A. Dönhöfer and D.N. Wilson, Tetracycline antibiotics and resistance mechanisms, Biol. Chem., 2014, 395, 559-575. [ Links ]
6 a) X.-Y. Xiao, D.K. Hunt, J. Zhou, R.B. Clark, N. Dunwoody, C. Fyfe, T.H. Grossman, W.J. O'Brien, L. Plamondon and M. Rönn, Fluorocyclines. 1. 7-Fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxy-tetracycline: a potent, broad spectrum antibacterial agent, J. Med. Chem., 2012,55,597-605; [ Links ] b) C. Sun, D.K. Hunt, R.B. Clark, D. Lofland, W.J. O'Brien, L. Plamondon and X.-Y. Xiao, Synthesis and antibacterial activity of pentacyclines: a novel class of tetracycline analogs, J. Med. Chem., 2011, 54, 3704-3731. [ Links ]
7 P.-E. Sum, A.T. Ross, P.J. Petersen and R.T. Testa, Synthesis and antibacterial activity of 9-substituted minocycline derivatives, Bioorg. Med. Chem. Lett., 2006,16, 400-403. [ Links ]
8 a) K. Nicolaou, C.R. Hale, C. Nilewski, H.A. Ioannidou, A. ElMar-rouni, L.G. Nilewski, K. Beabout, T.T. Wang and Y. Shamoo, Total synthesis of viridicatumtoxin B and analogues thereof: strategy evolution, structural revision, and biological evaluation, J. Am. Chem. Soc., 2014, 136, 12137-12160; [ Links ] b) C. Sun, D.K. Hunt, C.-L. Chen, Y. Deng, M. He, R.B. Clark, C. Fyfe, T.H. Grossman, J.A. Sutcliffe and X.-y.C. Xiao, Design, synthesis, and biological evaluation of hexacyclic tetracyclines as potent, broad spectrum antibacterial agents, J. Med. Chem., 2015; [ Links ] c) U. Lesnik, T. Lukezic, A. Podgorsek, J. Horvat, T. Polak, M. Sala, B. Jenko, K. Harmrolfs, A. Ocampo-Sosa and L. Martínez-Martínez, Construction of a new class of tetracycline lead structures with potent antibacterial activity through biosyn-thetic engineering, Angew. Chem. Int. Ed., 2015, 54, 3937-3940. [ Links ]
9 A. Ricci, Asymmetric Organocatalysis at the service of medicinal chemistry, ISRN. Org. Chem., 2014, 2014. [ Links ]
10 B. List, R.A. Lerner and C.F. Barbas, Proline-catalyzed direct asymmetric aldol reactions, J. Am. Chem. Soc., 2000,122, 2395-2396. [ Links ]
11 a) B. List, P. Pojarliev, W.T. Biller and H.J. Martin, The proline-catalyzed direct asymmetric three-component Mannich reaction: scope, optimization, and application to the highly enantioselective synthesis of 1, 2-amino alcohols, J. Am. Chem. Soc., 2002, 124, 827-833; [ Links ] b) Y. Xue, L.-P. Li, Y.-H. He and Z. Guan, Protease-catalysed direct asymmetric Mannich reaction in organic solvent, Sci. Rep., 2012, 2. [ Links ]
12 a) S. Sulzer-Mosse and A. Alexakis, Chiral amines as organocatalysts for asymmetric conjugate addition to nitroolefins and vinyl sulfones via enamine activation, Chem. Commun., 2007, 3123-3135; [ Links ] b) B. List, P. Pojarliev and H.J. Martin, Efficient proline-catalyzed Michael additions of unmodified ketones to nitro olefins, Org. Lett., 2001, 3, 2423-2425. [ Links ]
13 a) W. Notz, F. Tanaka and C.F. Barbas, Enamine-based organocatalysis with proline and diamines: the development of direct catalytic asymmetric Aldol, Mannich, Michael, and Diels-Alder reactions, Acc. Chem. Res., 2004, 37, 580-591; [ Links ] b) E. Zamani, Organocatalyzed stereospecific C-C bond formation of /?-lactams, Org. Biomol. Chem., 2013, 11, 8294-8297. [ Links ]
14 a) J. Alemán and S. Cabrera, Applications of asymmetric organocatalysis in medicinal chemistry, Chem. Soc. Rev., 2013, 42, 774-793; [ Links ] b) M. Raj and V.K. Singh, Recent advances on stereoselective organo-catalytic reactions. Organocatalytic synthesis of natural products and drugs, Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques, and Applications, 2011, 415. [ Links ]
15 a) Z.E. Cele, S.A. Pawar, T. Naicker, G.E. Maguire, P.I. Arvidsson, H.G. Kruger and T. Govender, Organocatalytic Mannich reactions on a carbapenem core - synthesis of Mannich bases and bicyclic diazano-nanes, Eur. J. Org. Chem., 2014, 2014, 2253-2260; [ Links ] b) Z.E. Cele, P.I. Arvidsson, H.G. Kruger, T. Govender and T. Naicker, Applied enantioselective aminocatalysis: a-heteroatom functionalization reactions on the carbapenem (S-lactam antibiotic) core, Eur. J. Org. Chem., 2015, 2015, 638-646; [ Links ] c) S. Khanyase, T. Naicker, G.E. Maguire, H.G. Kruger, P.I. Arvidsson and T. Govender, l-Proline organocatalyzed Michael synthesis of monobactam and carbapenem /?-lactam cores, Tetrahedron: Asymmetry, 2014, 25, 969-973. [ Links ]
16 S. Bala, N. Sharma, A. Kajal, S. Kamboj and V. Saini, Mannich Bases: An important pharmacophore in present scenario, Int. J. Med. Chem., 2014, 2014. [ Links ]
17 W. Siedel, A. Soder and F. Lindner, Die Aminomethylierung der Tetracycline. Zur Chemie des Reverin, Munch. Med. Wochenschr., 1958,17, 661-663. [ Links ]
18 K.M Huttunen and J. Rautio, Prodrugs - an efficient way to breach delivery and targeting barriers, Curr. Top. Med. Chem., 2011, 11, 2265-2287. [ Links ]
19 A.L. Simplício, J.M. Clancy and J.F. Gilmer, /?-Aminoketones as prodrugs with pH-controlled activation, Int. J. Pharm., 2007, 336, 208-214. [ Links ]
20 A. Cordova, The direct catalytic asymmetric Mannich reaction, Acc. Chem. Res., 2004, 37, 102-112. [ Links ]
21 P.I. Dalko, Enantioselective Organocatalysis, Wiley Online Library, 2007. [ Links ]
22 B.T. Hahn, R. Froehlich, K. Harms and F. Glorius, Proline-catalyzed highly enantioselective and anti-selective Mannich reaction of unactivatedketones: synthesis of chirala-amino acids, Angew. Chem. Int. Ed., 2008,47, 9985-9988. [ Links ]
23 a) M.M. Salter, J. Kobayashi, Y. Shimizu and S. Kobayashi, Direct-type catalytic three-component mannich reactions leading to an efficient synthesis of a, /?-diamino acid derivatives, Org. Lett., 2006, 8, 3533-3536; [ Links ] b) B. List, Proline-catalyzed asymmetric reactions, Tetrahedron, 2002, 58, 5573-5590. [ Links ]
24 a) C. Sun, Q. Wang, J. D. Brubaker, P.M. Wright, C.D. Lerner, K. Noson, M. Charest, D.R. Siegel, Y.-M. Wang and A.G. Myers, A robust platform for the synthesis of new tetracycline antibiotics, J. Am. Chem. Soc., 2008,130,17913-17927; [ Links ] b) M.L. Nelson, M.Y. Ismail, L. McIntyre, B. Bhatia, P. Viski, P. Hawkins, G. Rennie, D. Andorsky, D. Messersmith and K. Stapleton, Versatile and facile synthesis of diverse semisynthetic tetracycline derivatives via Pd-catalyzed reactions, J. Org. Chem.,2003, 68, 5838-5851. [ Links ]
25 Y. Takeuchi, Y. Imafuku and M. Nishikawa, Reassignment of the 13C NMR spectrum of minomycin, Arkivoc, 2003, 15, 39-46. [ Links ]
26 Clinical and Laboratory Standards Institute, Perfomance Standard for Antimicrobial Susceptibility Testing; Twenty-fourth Informational Supplement 34, 2014. [ Links ]
Received 22 January 2016
Revised 3 March 2016
Accepted 6 March 2016
* To whom correspondence should be addressed. E-mail: naickert1@ukzn.ac.za / govenderthav@icloud.com
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]


{kind=link}
{kind=link}
{kind=link}
{kind=link}