










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.69 Durban 2016
http://dx.doi.org/10.17159/0379-4350/2016/v69a5
RESEARCH ARTICLE
Lack of Co-crystal Formation with Cyclotriphosphazenes: A Cautionary Tale†
Helene Wahl; Delia A. Haynes*; Tanya le Roex
Department of Chemistry and Polymer Science, University of Stellenbosch, Private Bag XI, Matieland, 7602, South Africa
ABSTRACT
The attempted formation of co-crystals with a series of cyclotriphosphazene derivatives has been investigated. Despite numerous attempts, only one co-crystal was obtained. The crystal structure of this material, [hexakis(4-pyridyloxy)-cyclotriphos-phazene][terephthalic acid]2y is presented here. The crystal structures of 2,2-bis(4-formylphenoxy)-4,4,6,6-bis[spiro(2',2"-dioxy-1',1"-biphenylyl]cyclo-triphosphazene and hexakis(4-cyanophenoxy)cyclotriphosphazene are also reported for the first time. The extremely low rate of co-crystal occurrence in these materials cannot be explained, despite the consideration of several possibilities. This serves as a cautionary tale - co-crystal formation is not necessarily straightforward.
Keywords: Cyclophosphazene, lack of co-crystal formation
1. Introduction
Cyclophosphazenes, which are formally unsaturated compounds containing alternating phosphorus-nitrogen bonds,1 have received much attention due to the wide range of possible applications for these compounds.2 Cyclotriphosphazenes, (R2PN)3, have received particular attention from the crystal engineering community since the discovery of the remarkable inclusion properties of tris(o-phenylenedioxy)cyclotriphos-phazene (TPP).3 TPP is a highly robust host, forming inclusion complexes in the solid state with a variety of solvents, whether it is exposed to liquids or to solvent vapours.4 The high density, nonporous structure of TPP has also been determined, and it has been shown that under CO2 pressure the high density structure can be transformed to the low density, porous structure.5 Not only does this particular organic host include a wide variety of small organic molecules, but it has also been shown to include various polymers.6 We speculated that if TPP has the ability to form such a broad range of inclusion compounds, the possibility exists that other cyclotriphosphazene derivatives could also form a range of interesting supramolecular assemblies - be they inclusion compounds or co-crystals.7 It was therefore decided to embark on a systematic investigation of co-crystal and solvate formation with various cyclotriphosphazene derivatives.
Cyclotriphosphazenes are attractive targets for this kind of systematic study due to the ease of synthesis of a variety of substituted derivatives from hexachlorocyclotriphosphazene, (NPCl2)3.2a,4c,8 Despite this, there have been few systematic crystal engineering studies on cyclotriphosphazenes.9 Chandrasekhar et al.9aused cyclotriphosphazenes and the principles of directional hydrogen bonding to design supramolecular assemblies in the solid state. They specifically selected hydrazide derivatives with the intent of using the terminal -NH2 groups as proton donors and the phosphazene ring nitrogen atoms as proton acceptors. This interaction results in the formation of a hexagonal close-packed sheet in the case of N3P3[N(Me)NH2]6. When one of the phosphorus atoms is substituted with a 2,2'-biphenol group - spiro-N3P3[O2C12H8][N(Me)NH2]4-it results in the formation of a double chain through the hydrogen bonding of a ring nitrogen atom to a N(Me)NH2 substituent. This is one of the few examples of using tailored intermolecular interactions to direct the aggregation of cyclotriphosphazenes.
There are also a number of known co-crystals containing a cyclotriphosphazene derivative.10 One of the most interesting co-crystals is that of hexakis(4-carboxyphenoxy)-cyclo-triphosphazene with hexakis(4-pyridylcarbinoxy)cyclo-triphosphazene (CSD refcode EZEJEY).11,12 Hydrogen bonds are formed between the carboxylic acid and pyridyl groups on the cyclotriphosphazenes. Due to the three-up and three-down arrangement of the substituents on each cyclotriphosphazene, they assemble to form columns in the crystal structure. Halogen bonding between iodo-substituted perfluorocarbons and pyridyl moieties has been successfully used to construct co-crystals of cyclotriphosphazenes.10a,b One of these co-crystals also forms a pillared structure due to the arrangement of the cyclotriphosphazene substituents.10b These co-crystals served as the inspiration to design similar pillared assemblies with different cyclotriphosphazene derivatives.
We selected a number of substituted cyclotriphosphazenes (Scheme 1) that have either the potential to form strong supra-molecular synthons (hydrogen or halogen bonds), or have awkward shapes, or both. We hoped that the flexibility and awkward shape of these molecules in general might predispose them to crystallize as solvates in the solid state. The co-formers that were selected had the potential to form known supramo-lecular synthons with the functional groups on the phosphazenes (hydrogen bonds, halogen bonds, π-π interactions).
At the start of this investigation, it was anticipated that several co-crystals would be obtained. Molecules and co-crystal formers with the potential to form strong supramolecular synthons were selected, and a thorough screen was planned. In general, publications involving co-crystal synthesis do not report problems encountered in obtaining co-crystals. A close look at the most recent papers in a top crystal engineering journal13 containing the word co-crystal (or cocrystal) in the title confirms this. One of the studies discusses salt versus co-crystal formation, and it appears multi-component crystals were obtained for all combinations investigated.13c Four of the studies involve co-crystal screens. In one screen it seems all combinations tried gave co-crystals.13a In the other three screens, co-formers were chosen so as to form strong intermolecular interactions. Two screens had low rates of co-crystal formation (1 co-crystal from 8 combinations13b and 4 co-crystals from 25 combinations13e), but make no comment on this. The third screen found 4 co-crystals from around 300 crystallizations.13d The researchers comment 'The scarcity of co-crystals of fenamic acid derivatives, as reported in this study and in previous literature, is due to both the strong carboxylic acid homodimer synthon that is dominant within the numerous polymorphs of the fenamic acid derivatives, and the ability for the molecules to adopt several stable conformations of their own. The strength of these interactions within these systems results in the evident preferential formation of poly-morphs over co-crystals.'
Some groups have reported investigations into why certain co-crystals (or groups of co-crystals) do not readily form; however, these papers are few and far between.14 From the literature, a newcomer to this field might expect that obtaining a co-crystal is a relatively simple matter - as long as an appropriately strong interaction could form between the two co-crystal formers, a co-crystal will be isolated. The results of our extensive study, reported herein, show that this is most certainly not always the case.
2. Experimental
Cyclophosphazenes 1-12 were synthesized using established literature procedures, with some adaptations where necessary. Full details are given in the Supplementary material.
Almost 300 co-crystallization experiments were carried out in this study. Details of these experiments are also given in full in the Supplementary material. The choice of co-former was based on the potential to form strong supramolecular synthons. For example, phosphazenes containing pyridyl substituents were co-crystallized with molecules containing carboxylic acids or hydroxyl groups, in the hope that formation of a pyridyl...HO hydrogen bond would drive co-crystal formation. We also investigated co-formers that might form π-π interactions or halogen bonds. Finally, bulky or awkwardly-shaped molecules were also investigated as co-formers. For lists of co-crystal formers and solvates used, see Tables S1-S17 in the Supplementary material. Very few mechanochemical experiments were carried out, as this tended to result in degradation of the phosphazene ring, most likely forming phosphazene polymers. There were no issues with solubility of the phosphazenes used.
For the structures of 1, 5, and 10-12, X-ray intensity data were collected on a Bruker-Nonius SMART Apex diffractometer equipped with a fine-focus sealed tube and a 0.5 mm Monocap collimator (monochromated Mo-Ka radiation, 1 = 0.71073 A). Data were captured with a CCD area-detector with the generator powered at 40 kV and 30 mA. The temperature of the crystal was controlled by constant stream of nitrogen gas produced by an Oxford Cryosystems Cryostat (700 Series Cryostream Plus). For the crystal structures of 6 and the co-crystal 7'TPA, intensity data were collected on a Bruker Apex DUO CCD diffractometer with a multilayer monochromator. Mo-Ka radiation (1 = 0.71073 A) was selected for the experiments. The temperature of the crystal was controlled using an Oxford Cryosystems Cryostream. Data reduction was done by means of a standard procedure using the Bruker software package SAINT15 and the absorption corrections and the correction of other systematic errors were performed using SADABS.16 The structures were solved by direct methods using SHELXS-97 and refined using SHELXL-97 and SHELXL-2014/7.17 X-Seed18 was used as the graphical interface for the SHELX program suite. Most hydrogen atoms were placed in calculated positions using riding models. Hydrogen atoms on nitrogen or oxygen atoms were located in the electron density difference map and allowed to refine.
Cambridge Structural Database12 (CSD) searches were carried out using ConQuest19 version 1.17 on version 5.36 with 3 updates (Nov. 2014, Feb. 2015, May 2015).
For powder X-ray diffraction analysis, samples were ground to a fine powder with a mortar and pestle and placed on a zero-background sample holder. Experiments were carried out on a PANalytical X'Pert PRO instrument in Bragg-Brentano geometry. Intensity data were collected using an X'Celerator detector and 20 scans in the range of 5-50° were performed using Cu-Ka radiation (λ= 1.5418 A).
Differential scanning calorimetry was carried out using a TA Instruments Q100 system under an N2 gas purge, with a flow rate of 50.0 ml min-1. The samples (ranging from 1.5 to 3 mg) were placed in aluminium pans that were non-hermetically sealed with vented aluminium lids. The heating rate for all experiments was 10 °C min-1, and the cooling rate was 5 °C min-1.
3. Results and Discussion
3.1. CSD Analysis
In order to give some idea of the frequency with which cyclotriphosphazenes form co-crystals or solvates, a Cambridge Structural Database search was carried out.12 If the search is restricted to 'organic only' structures with 3D co-ordinates determined, 728 structures containing the cyclotriphosphazene moiety are identified. Of these, 26 are salts or zwitterions (identified by searching for any charged atom). These were removed (as salts are necessarily multi-component crystals). Of the remaining 702 structures, only 109, or 15.5 %, are multi-component crystals (>1 chemical residues). This is only slightly lower than the occurrence in the CSD as a whole, where 16.8 % (42 601/252 827) of non-ionic organic structures are multi-component crystals.
These results imply that co-crystal or solvate formation is no more or less likely to occur with cyclotriphosphazenes than with any other molecule.
3.2. Co-crystallization Experiments
3.2.1. Co-crystallizations with 1
The choice of 1 as a potential co-crystal former was based on its paddlewheel conformation, which indicated that it may form inclusion compounds similar to those formed by TPP.3,4,5,6 The NH-functional groups on 1 are also available as potential hydrogen bond donors. A number of co-crystallization experiments were carried out with 1 (Table S1). Only the crystallization from THF/hexane yielded crystals. These proved to be the
known hydrate of 1 (CSD refcode COPVAE).20 We have re-determined this structure at 173 K, and crystallographic data are given in Table 1. El Murr et al.20report the isolation of crystals of non-solvated 1 from the reaction mixture, but these crystals did not survive exposure to X-rays. However, the crystals grown from methanol that incorporated water into the structure did not disintegrate upon exposure to X-rays, suggesting that incorporation of water increases the stability of the crystal. It seemed likely that other hydrogen bonding solvents might also fulfil this role, and we therefore attempted crystallization from a number of hydrogen bonding solvents (donors and acceptors, see Table S1) in the hope of forming solvates. This was unsuccessful.
3.2.2. Co-crystallization Experiments with 2, 3 and 4
Numerous attempts to generate either solvates or co-crystals with 2,3 and 4 were made (see Tables S2, S4 and S6 in the Supplementary material). None of these experiments yielded multi-component crystals. In most cases either no crystals were obtained, or crystals of the starting materials were obtained.
In order to investigate whether additional energy might be required for co-crystal formation, mechanochemical experiments were carried out with 2, 3 and 4. These derivatives were ground together with 4,4'-bipyridine, imidazole or benzimidazole (Tables S3, S5, S7). The products obtained on grindingboth 2 and 3 with imidazole were sticky white substances, whereas all the other products were fine dry powders. The products of the grinding experiments were analysed by PXRD, which showed that all products consisted of physical mixtures of the reagents (see Supplementary material).
The product obtained by grinding 2 with benzimidazole was dissolved in THF. This yielded a pink precipitate with a powder pattern that differs from those of both 2 and benzimidazole. NMR analysis of this precipitate showed numerous signals in the 31P spectrum that do not correspond with those normally observed for 2. This could indicate that grinding the two components together resulted in substitution of the chloro groups on the cyclotriphosphazene ring, or that the cyclotriphosphazene ring has opened and polymerized. The material is clearly not a co-crystal of 2.
3.2.3. Crystal Structures of 5 and 6
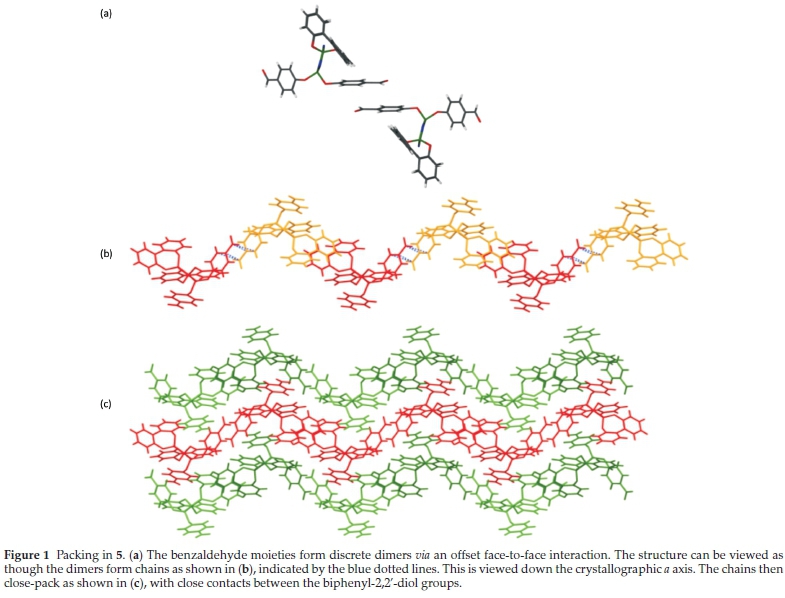
Co-crystallization experiments were carried out with 5 in the expectation that the awkward shape of the molecule might cause inefficient packing in the solid state, resulting in inclusion of solvent molecules. Attempts were therefore made to crystallize 5 from a variety of solvents (Table S8). This yielded crystals of 5 in two cases (benzene and acetone). As the crystal structure of this material has not previously been reported, it is described here. Selected crystallographic data are given in Table 1.
Compound 5 crystallizes in the monoclinic space group P21/n. One of the benzaldehyde groups is disordered over two positions of approximately equal occupancy. The molecules form discrete dimers via an offset face-to-face interaction between benzaldehyde groups, and the dimers are arranged in chains with p-stacking interactions between the biphenyl groups (Fig. 1). There are no hydrogen bonds evident in the structure, confirming that the aldehyde moiety is not a particularly strong hydrogen bond acceptor. Despite this, 5 was co-crystallized with a wide range of hydrogen bond donors. These co-crystallization experiments are listed in Table S9 in the Supplementary material. The ratio of 5 to co-crystal former was 1:2 in most experiments in order to take into account the two aldehyde moieties that could potentially participate in hydrogen bonding interactions. None of these experiments yielded any co-crystals. Some crystallizations produced crystals of 5, some produced crystals of the co-formers and some yielded no crystals at all.
Compound 6 was chosen for its potential to form supramolecular synthons through the nitrile functionality. It is sparingly soluble in most solvents, except for DMSO and DMF in which it readily dissolves. Several co-crystallization experiments were carried out with halogen-containing co-crystal formers (see Table S10); however, no co-crystals were obtained. Rod-shaped crystals of pure 6 were grown from a co-crystallization experiment with bromopentafluorobenzene in DMSO. Selected crys-tallographic details are given in Table 1.
Cyclophosphazene 6 crystallizes in P-1, with one molecule in the asymmetric unit. Two cyanophenyl moieties are approximately parallel to one another above the plane of the phospha-zene ring, and another two below the plane of the ring are also approximately parallel to one another. The other two cyano-phenyl moieties are twisted away from one another (Fig. 2a). The molecules stack on top of one another to give columns parallel to the crystallographic c axis (Fig. 2b). These columns pack alongside one another in a slightly offset fashion. There are no immediately apparent structure-directing intermolecular interactions in this structure.

3.2.4. Co-crystallization Experiments with 7 and 8
Both 7 and 8 were chosen as possible co-crystal formers because of the potential for hydrogen bond formation with either the pyridyl or the hydroxyl functionality on the molecules. One of the few known cyclotriphosphazene co-crystals not based on TPP contains hexakis(4-pyridylcarbinoxy)cyclo-triphosphazene, which is closely related to 7 (although the 3-D coordinates for this co-crystal structure are not in the CSD). Compound 7 therefore seemed a particularly promising candidate for co-crystal formation.
Numerous co-crystallization experiments were carried out with both 7 and 8 (Tables S11 and S12 in the Supplementary material). No co-crystals containing 8 were isolated. However, 7 was found to form a co-crystal with terephthalic acid, 7'TPA. Crystallographic data are given in Table 1.
The crystals of 7'TPA were grown from a 1:2 ratio of 7:terephthalic acid in a layered solution of chloroform and DMF. Crystals of 7 were dissolved in chloroform and the terephthalic acid in DMF. Care had to be taken to add the two solutions together slowly as terephthalic acid is insoluble in chloroform and precipitates out of solution when chloroform is added to DMF. The co-crystal, which contains 2.5 terephthalic acid molecules for every molecule of 7, crystallizes in P-1. No charge transfer has taken place, i.e. both carboxylic acid groups of each terephthalic acid molecule are protonated, and these form hydrogen bonds with the pyridyl groups on the cyclotriphos-phazene. Five of the six pyridyl groups on the cyclotriphos-phazene ring are involved in hydrogen bonding with a terephthalic acid molecule through a COOH---N interaction with the hydroxyl group of the carboxylic acid. This results in an infinite hydrogen-bonded chain (Fig. 3a). The terephthalic acid molecules shown in orange in Fig. 3b link neighbouring chains through hydrogen bonding to a pyridyl group in the next chain.

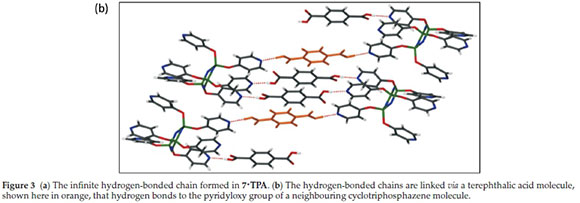
It seems likely that the hydrogen bonds between 7 and terephthalic acid are strong interactions, driving the formation of a co-crystal. It is surprising, however, that no other co-crystals with di-acids were isolated with this derivative despite this seemingly strong interaction. This could indicate that the correct shape match is very important in forming co-crystals with cyclotriphosphazenes.
3.2.5. Co-crystallization Experiments with 9-12
The hexakis(4-halophenoxy)cyclotriphosphazene derivatives 9-12 were synthesized in order to investigate whether co-crystals could be formed using supramolecular synthons involving halogens. These derivatives were co-crystallized with a range of small molecules, including molecules containing halogens, nitriles and nitrogen-containing heterocycles such as pyridine. The solvent systems that were used include acetonitrile, acetonitrile/DCM, DCM, THF, THF/methanol, chloroform, chloroform/acetone and DMSO, as well as combinations of these solvents (Tables S13-16). In each case the cyclotriphosphazene derivative crystallized as the pure material, rather than co-crystallizing with other small molecules.
The crystal structures of 9-12 were originally determined at room temperature.20 Our previous observation of polymorphism in 921 led us to redetermine the structures at 100 K, and the data are included here for completeness (see Table 1). On analysis of the crystal structures of these derivatives, it is apparent that hexakis(4-fluorophenoxy)cyclotriphosphazene (9) and hexa-kis(4-chlorophenoxy)cyclotriphosphazene (10) are isostruc-tural, and that hexakis(4-bromophenoxy)cyclotriphosphazene (11) and hexakis(4-iodophenoxy)cyclotriphosphazene (12) are isostructural. The only differences between the structures are slight changes in the twist of the phenoxy rings between derivatives. DSC was also carried out on 9-12, but only 9 showed any evidence of polymorphism in the temperature range investigated.
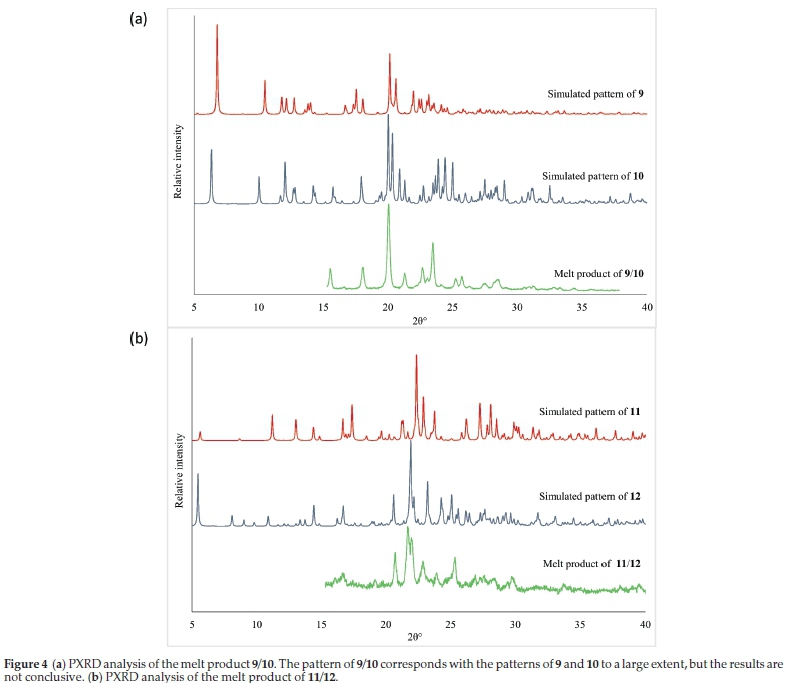
The isostructurality between 9 and 10, as well as 11 and 12, implied that combinations of these molecules may lead to co-crystal formation. Unfortunately, co-crystals of 9 with 10,or 11 with 12, could not be grown from solution. The growth of co-crystals by melting the isostructural compounds together was then investigated (Table S17). Crystals of 9 and 10 were ground together in a 1:1 ratio, melted and DSC analysis was performed on the product. DSC analysis showed that the melting point of the product (referred to as 9/10) is at 122.9 °C, which is lower than the melting point of both 9 (129 °C) and 10 (152 °C). The same procedure was followed for 11 and 12, and again there is only one melting point at 173.1 °C. The melting point of 11 is 176.8 °C and that of 12 is 187.6 °C. This is, however, not conclusive evidence that a co-crystal has formed.
PXRD analysis was carried out on the melt products 9/10 and 11/12. The results are shown in Fig. 4. PXRD analysis is unfortunately also not conclusive. In both cases, the powder patterns of the melt products largely correspond to the simulated patterns of the halophenoxy derivatives. There are minor differences between the patterns, but it is difficult to determine whether a novel co-crystal has in fact formed. It is likely that any co-crystals formed would be isostructural to the co-crystal formers, and hence have similar powder patterns. The melt products could also be solid solutions, as opposed to stoichiometric co-crystals. We are thus unable to state conclusively whether these derivatives form co-crystals from the melt.
3.3. Lack of Co-crystal Formation
During this investigation, a total of nearly 300 co-crystallization experiments were carried out using 12 different cyclophosphazene derivatives and a number of crystallization techniques, and only one co-crystal was obtained. Largely, the crystals that were obtained were of the pure cyclotriphos-phazene derivative, with only one derivative (1) crystallizing as a hydrate and no derivatives crystallizing as solvates. This is a remarkably low success rate - the CSD study indicates that around 15 % of cyclotriphosphazene-containing structures are multi-component crystals. Why then was our success rate so low?
The most obvious explanation is that the supramolecular synthons employed were simply not strong enough to promote co-crystallization, i.e. the heteromeric interactions were not as strong as the homomeric interactions. This may well have been the case for many of the combinations that were attempted, although in a number of the molecules selected there are no obvious strong intermolecular interactions in the crystals of the pure material. However, this does not explain why a co-crystal could be formed between 7 and terephthalic acid, but not between 7 and any other carboxylic acids, despite numerous attempts.
Inspection of the pKa values of the acids used (Table 2)22 indicates that this is not simply a case of acid strength (and by extension hydrogen bond strength). There are clearly other factors involved.
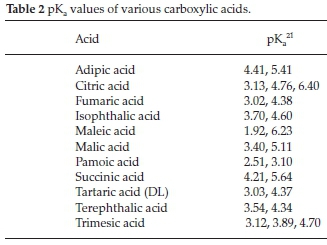
It does appear that the size and shape of the co-crystallization agent plays a role. A study conducted by Anderson et al.23found that molecules that crystallize with Z'>1are generally small, awkwardly shaped molecules. They are also more conformationally restricted, which might explain why the cyclotriphosphazenes seldom co-crystallize with smaller molecules: cyclotriphosphazenes are much larger molecules with substituents that are more conformationally flexible. It has also been suggested that polymorphic molecules make better co-crystal formers.24 We do not observe this: compound 9 is highly polymorphic, with four known polymorphs, yet no co-crystals of this molecule were isolated. In this case, the conformational flexibility of the molecule results in it crystallizing in a number of ways - the molecule can solve the problem of close-packing without the need for a co-former. In order to improve the probability of obtaining co-crystals or solvates with cyclotriphosphazenes, the substituents on the P-N ring should be conformationally restricted (no flexible O linkage). Functional groups on the substituents should be strong hydrogen bond donors or acceptors, or strong halogen bond acceptors (such as iodine where the acceptor strength has been increased by fluorination).
This study aimed to isolate co-crystals from solution, in the manner one might carry out a co-crystal screen for a pharmaceutical molecule. The difficulty encountered in obtaining co-crystals was not at all anticipated. It is possible that with further detailed investigation, the particular conditions to form co-crystals of these molecules could be identified. Several avenues were pursued to try and explain the low success rate, including analysis of what preferred co-crystal formers might be. None of these offered any insight - in fact, all analysis indicated that we should be able to form other co-crystals with 7.Itis particularly interesting that no further co-crystals between 7 and carboxylic acids were obtained. No obvious explanation for this can be identified.
4. Conclusions
An extensive investigation of co-crystal formation with 12 cyclotriphosphazene derivatives has been carried out. Around 300 co-crystallization experiments were carried out, but only one co-crystal was obtained. These results clearly indicate that many cyclotriphosphazenes have a very low tendency to form co-crystals. There is no clear explanation for this, but it serves as a caution: the formation of co-crystals is not necessarily trivial, and certainly more investigation is needed to shed light on why this is the case.
Acknowledgements
We thank the National Research Foundation* of South Africa, Stellenbosch University and the SU Department of Chemistry and Polymer Science for funding.
Supplementary Material
Details of co-crystallization experiments, PXRD, NMR, DSC and synthetic details are available as supplementary information. X-ray crystallographic data are available in cif format. CCDC 1055282-1055288 contain the crystallographic data for this manuscript. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References and Notes
1 N.N. Greenwood and A. Earnshaw, Chemistry of the Elements, 2nd edn., Pergamon Press, Great Britain, 1997. [ Links ]
2 (a) M. Gleria and R.J. De Jaeger, Aspects of phosphazene research, J. Inorg. Organomet. Polym., 2001, 11, 1-45. [ Links ] (b) R. De Jaeger and M. Gleria, Poly(organophosphazene)s and related compounds: synthesis, properties and applications, Prog. Polym. Sci., 1998, 23, 179-276. [ Links ]
3 H.R. Allcock, Phosphonitrilic compounds. II. Reactions of phospho-nitrilic chlorides with catechol and triethylamine, J. Am. Chem. Soc., 1964, 86, 2591-2595. [ Links ]
4 (a) H.R. Allcock, R.W. Allen, E.C. Bissell, L.A. Smeltz and M. Teeter, Phosphorus-nitrogen compounds. 26. Molecular motion and molecular separations in cyclophosphazene clathrates, J. Am. Chem. Soc., 1976, 98, 5120-5125. [ Links ] (b) H.R. Allcock, M.L. Levin and R.R. Whittle, R.R., Tris(o-phenylenedioxy)cyclotriphosphazene: the clathration-induced monoclinic to hexagonal solid-state transition, Inorg. Chem., 1986, 25, 41-47. [ Links ] (c) G. Couderc and J. Hulliger, Channel forming organic crystals: guest alignment and properties, Chem. Soc. Rev., 2010, 39, 1545-1554. [ Links ]
5 J. Tian, P. Thallapally, J. Liu, G.J. Exarhos and J.L. Atwood, Gas-induced solid state transformation of an organic lattice: from non-porous to nanoporous, Chem. Commun., 2011, 47, 701-703. [ Links ]
6 (a) H.R. Allcock, A.P. Primrose, N.J. Sunderland, A.L. Rheingold, I.A. Guzei and M. Parvez, Inclusion of polymers within the crystal structure of tris(o-phenylenedioxy)cyclotriphosphazene, Chem. Mater., 1999,11,1243-1252. [ Links ] (b) S. Bracco, A. Comotti, P. Valsesia, M. Beretta and P. Sozzani, Self-assembly of 1,4-cis-polybutadiene and an aromatic host to fabricate nanostructured crystals by CH...r interactions, CrystEngComm, 2010,12, 2318-2321. [ Links ]
7 We take co-crystal to imply any multi-component crystal where the components occur in a stoichiometric ratio, and are neutral. Solvates are included in this definition, but we refer to what are conventionally called solvates as such to avoid confusion. [ Links ]
8 (a) H.R. Allcock, Recent advances in phosphazene (phosphonitrilic) chemistry, Chem. Rev., 1972,72,315-356; [ Links ] V. Chandrasekhar, P. Thilagar and B.M. Pandian, Cyclophosphazene-based multi-site coordination ligands, Coord. Chem. Rev., 2007,251,1045-1074. [ Links ] (b) A. Steiner, Supra-molecular structures of cyclotriphosphazenes, in Polyphosphazenes for Biomedical Applications, John Wiley & Sons, 2008, pp. 411-453. [ Links ]
9 (a) V. Chandrasekhar, V Krishnan, G.T.S. Andavan, A. Steiner and S. Zacchini, Cyclophosphazene supramolecular assemblies: N-H--N and C-H--N mediated supramolecular networks in the crystal structures of N3P3[N(Me)NH2]6 and spiro-N3P3[O2C12H8][N(Me)NH2]4, CrystEngComm. 2003,5,245-247. [ Links ] (b) V. Chandrasekhar, P. Thilagar, V Krishnan, J.F. Bickley and A. Steiner, Click synthesis of fluorine-rich cyclotriphosphazene hydrazones. synthesis and supramolecular structures of N3P3(N(Me)N = CHC6F5)6, spiro-N3P3(C12H8O2) (N(Me)N=CHC6F5)4, and dispiro-N3P3(C12H8O2)2(N(Me)N = CHC6F5)2, Cryst. Growth Des., 2007, 7, 668-675. [ Links ]
10 (a) R. Bertani, E. Ghedini, M. Gleria, R. Liantonio, G. Marras, P. Metrangolo, F. Meyer, T. Pilati and G. Resnati, Cyclotriphosphazene [N3P3(2,2'-dioxybiphenyl)2-(4-pyridinoxy)2] and its halogen bonded complex with 1,4-diiodotetrafluorobenzene, CrystEngComm, 2005, 7, 511-513. [ Links ] (b) R. Bertani, F. Chaux, M. Gleria, P. Metrangolo, R. Milani, T. Pilati, G. Resnati, M. Sansotera and A. Venzo, Supramolecular rods via halogen bonding-based self-assembly of fluorinated phosphazene nanopillars, Inorg. Chim. Acta, 2007, 360, 1191-1199. [ Links ] (c) T. Itaya, N. Azuma and K. Inoue, Self-Assembly of hexakis(4-pyridyl-methoxy)cyclotriphosphazene and 1,4-anthracenedicarboxylic acid: structure and inclusion behavior, Bull. Chem. Soc. Jpn., 2002, 75, 2275-2281. [ Links ]
11 K. Inoue, T. Itaya and N. Azuma, Self-assembly through hydrogen bonding of cyclotriphosphazenes. Formation of cylindrical structures, Supramol. Sci., 1998, 5, 163-166. [ Links ]
12 F.H. Allen, The Cambridge Structural Database: a quarter of a million crystal structures and rising, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002, 58, 380-388. Reference 11 reports on interactions in the crystal structure. There are however no 3D-coor-dinates for this structure in the Cambridge Structural Database. [ Links ]
13 Search was carried out on 31 August 2015 in CrystEngComm. A total of eleven papers with a 2015 date were identified. Six of these discuss only a single new co-crystal, with no screening carried out. These are not included here. (a) A. Jacobs and F.M. Amombo Noa, Co-crystals and co-crystal hydrates of vanillic acid, CrystEngComm, 2015, 17, 98-106. [ Links ] (b) Y. Yan, J.-M. Chen and T.-B. Lu, Thermodynamics and preliminary pharmaceutical characterization of a melatonin-pimelic acid cocrystal prepared by a melt crystallization method, CrystEngComm, 2015, 17, 612-620. [ Links ] (c) A. Lemmerer, S. Govindraju, M. Johnston, X. Motloung and K.L. Savig, Co-crystals and molecular salts of carboxylic acid/pyridine complexes: can calculated pKa's predict proton transfer? A case study of nine complexes, CrystEngComm, 2015, 17, 3591-3595. [ Links ] (d) K. E. Wittering, L. R. Agnew, A. R. Klapwijk, K. Robertson, A.J.P. Cousen, D.L. Cruickshank and C.C. Wilson, Crystallisation and physicochemical property characterisation of confor-mationally-locked co-crystals of fenamic acid derivatives, CrystEngComm, 2015, 17, 3610-3618. [ Links ] (e) A. S. Sinha, U. B. Rao Khandavilli, E.L. O'Connor, B.J. Deadman, A.R. Maguire and S.E. Lawrence, Novel co-crystals of the nutraceutical sinapic acid, CrystEngComm, 2015, 17, 4832-4841. [ Links ]
14 See for example (a) N. Báthori, A. Lemmerer, G.A. Venter, S.A. Bourne and M.R. Caira, Pharmaceutical co-crystals with isonicoti-namide-vitamin B3, clofibric acid, and diclofenac - and two isonicotinamide hydrates, Cryst. Growth Des., 2011, 11, 75-87. [ Links ] (b) D.-K. Buçar, G.M. Day, I. Halasz, G.G.Z. Zhang, J.R.G. Sander, D.G. Reid, L.R. MacGillivray, M.J. Duer and W. Jones, The curious case of (caffeine)-(benzoic acid): how heteronuclear seeding allowed the formation of an elusive co-crystal, Chem. Sci., 2013, 4, 4417-4425. [ Links ] (c) M.A. Solomos, C. Mohammadi, J.H. Urbelis, E.S. Koch, R. Osborne, C.C. Usala and J.A. Swift, Predicting cocrystallization based on heterodimer energies: the case of Ν,Ν'-diphenylureas and triphenyl-phosphine oxide, Cryst. Growth Des., 2015,15, 5068-5074. [ Links ]
15 SAINT Data Reduction Software, Version 6.45; Bruker AXS Inc., Madison, WI, 2003. [ Links ]
16 (a) SADABS, Version 2.05; Bruker AXS Inc., Madison, WI, 2002. [ Links ] (b) R. H. Blessing, An empirical correction for absorption anisotropy, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995,51, 33-38. [ Links ]
17 G.M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112-122. [ Links ]
18 L.J. Barbour, X-Seed-A software tool for supramolecular crystallography, J. Supramol. Chem., 2001,1, 189-191. [ Links ]
19 I.J. Bruno, J. C. Cole, P.R. Edgington, M. Kessler, C.F. Macrae, P McCabe, J. Pearson and R. Taylor, New software for searching the Cambridge Structural Database and visualising crystal structures, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002,58,389-397. [ Links ]
20 N. El Murr, R. Lahana, J.-F. Labarre and J.-P. Declercq, An answer to the spiro versus ANSA dilemma in cyclophosphazenes: Part V. The dispiro N3P3Cl2 [HN-(CH2)3,4-NH]2 and trispiro N3P3 [HN-(CH2)3-NH]3 derivatives, J. Mol. Struct., 1984,117, 73-85. [ Links ]
21 H. Wahl, D.A. Haynes and T. le Roex, A series of polymorphs of hexakis(4-fluorophenoxy)cyclotriphosphazene, Cryst. Growth Des., 2012, 12, 4031-4038. [ Links ]
22 (a) D.R. Lide, Editor-in-Chief, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 88th edn, 2008. [ Links ] (b) H.C. Brown, D.H. McDaniel and O. Háfliger, in Determination of Organic Structures by Physical Methods, (E.A. Braude and F.C. Nachod, eds.), Academic Press, New York, 1955. [ Links ] (c) Handbook ofPharmaceutical Salts: Properties, Selection and Use, (P.H. Stahl and C.G. Wermuth, eds.), Wiley-VCH/VHCA, Weinheim/Zürich, 2002. [ Links ]
23 K.M. Anderson, M.R. Probert, A.E. Goeta and J.W. Steed, Size does matter-the contribution of molecular volume, shape and flexibility to the formation of co-crystals and structures with Z' > 1, CrystEngComm, 2011, 13, 83-87. [ Links ]
24 C.B. Aakeröy and D.J. Salmon, Building co-crystals with molecular sense and supramolecular sensibility, CrystEngComm, 2005, 7, 439-448. [ Links ]
Received 13 November 2015
Revised 27 January 2016
Accepted 1 February 2016
† We dedicate this paper to the many postgraduate students we have met over the past few years who despair of ever getting a co-crystal. You are not alone.
* To whom correspondence should be addressed. E-mail: dhaynes@sun.ac.za
This material is based upon work supported financially by the National Research Foundation of South Africa. Any opinion, findings and conclusions or recommendations expressed in this material are those of the authors and therefore the NRF does not accept any liability in regard thereto.
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]


{kind=link}
{kind=link}
{kind=link}
{kind=link}