










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.68 Durban 2015
http://dx.doi.org/10.17159/0379-4350/2015/V68A12
RESEARCH ARTICLE
Evaluation of six sample preparation methods for determination of trace metals in lubricating oils using inductively coupled plasma-optical emission spectrometry
Haile A. TekieI; Robert I. McCrindleI, *; Pieter J.J.G. MaraisI; Abayneh A. AmbusheII
IDepartment of Chemistry, Tshwane University of Technology, P.O Box 56208, Arcadia, Pretoria, 0007, South Africa
IIDepartment of Chemistry, University of Limpopo, Private Bag X1106, Sovenga, 0727, South Africa
ABSTRACT
Quantification of trace elements in used lubricating oil forms a vital part in monitoring engine conditions and impact on the environment. In this study, inductively coupled plasma-optical emission spectrometry (ICP-OES) was employed for the determination of Ag, Ba, Cu, Mn and Ni in used lubricating oils. Methodology was developed so as to minimize the oil's carbonaceous matter and its effect on viscosity. Accordingly, six oil sample preparation techniques (xylene dilution, detergent emulsion, microwave digestion, dry-ashing, wet-ashing and ultrasonic extraction) were investigated for their efficiency. Optimization of the factors influencing ultrasonic-assisted extraction and ICP-OES operating parameters enabled quantification of the trace metals in oils. Limits of detection (3Sb/m), in the ng g-1 range, were obtained for each element of interest using each method investigated. The validity of the methodologies studied was confirmed through the analysis of quality control (QC) samples. Analyte recoveries, ranging from 48.3 to 106 %, were obtained. Evaluation of the analytical methods studied with regard to accuracy, precision, LOD, linearity, applicability for routine analysis, preparation time and cost was made. Based on these evaluations, ultrasonic extraction has a clear advantage in terms of accuracy, applicability for routine analysis, time and cost of sample preparation.
Keywords: Lubricating oil, ICP-OES, optimization
1. Introduction
Trace metals in lubricating oils are generated from additive metals, wear particles1-3 and contaminants.2-4 Premium quality base oils are usually blended with organo-metallic compounds of Ba, Ca, Mg,1,5,6 Mo, P, Zn,1,5 Cd, Co, Cr, Fe, Hg, Ni, S, Sb, Se, Sn and Ti to enhance the performance of the lubricant and its properties.1 Mechanical erosion of moving parts usually introduces wear metals into the oil circulation.3 These wear products are composed of the same material as the metal surfaces from which they originated. During normal operation, wearing is inevitable. However, excessive friction causes premature wear, which results in significant economic loss.2,7 Any unwanted substance ingress into the oil is considered a contaminant. The most common oil contaminants include: particle, sand, moisture, dust, glycol, slag, soot and fuel.4 A recent study by Nissan motors of Japan reported that 85 % of hydraulic failures were due to oil contamination.4,8
Quantitative determination of trace metals in used oil serves as a diagnostic tool for engine and turbine faults.1,7 Currently, regular oil testing forms an integral part of a maintenance plan for shipping, aviation, oil refineries, mining, processing and chemical plants.8 Used oil analysis is thus used to perform preventive maintenance,4,6,7 replacement of components,7 predictive maintenance4,5 and control the quality of oil additives.5 However, lubricating oil analysis is often a difficult task due to its matrix complexity, viscosity and high organic content.1,5,6,9 Hence, analytical techniques with greater sensitivity are required for oil analysis. Spectrometric techniques such as flame-atomic absorption spectrometry (F-AAS), graphite furnace-atomic absorption spectrometry (GF-AAS) and inductively coupled-optical emission spectrometry (ICP-OES) have been widely used for oil analysis.10 In this report, ICP-OES was selected for its good limit of detection (LOD), better precision and repeatability, wider linear dynamic range7 and sequential multi-element analysis.11
Most analytical techniques require the sample for analysis to be in solution form. This solution should not contain high levels of insoluble particles and it should not be viscous.2 Samples with high organic load affect the plasma stability, increase background emission and reduce the energy suitable for ionization and excitation.7 Unfortunately, most preparation methods for oils are labour-intensive and time-consuming, particularly for large sample numbers.2 Consequently, 60 % of the total time required for analysis is spent in sample preparation and this accounts for 30 % of the total analysis error.12,13
Several oil sample preparation techniques are proposed in the literature, including: oil dilution, oil-in-water emulsification, microwave digestion,1,2,5,7,14 dry-ashing,1,2,5,14 wet-ashing2,6 and ultrasound-assisted extraction.15 The traditional oil dilution, using organic solvents, is convenient for rapid estimation of indicator metals. Xylene and kerosene have been widely used as typical solvents with ICP-OES.2 Oil-in-water emulsion, which employs a surfactant, converts the oil sample into homogenously dispersed micro-droplets in an aqueous phase. A homogenously dispersed emulsion behaves similar to an aqueous solution and allows aqueous standards for calibration.1,6,7,11 Microwave digestion is carried out in closed and pressurized vessels, employing various acids or acid mixtures.16 Digestion, using nitric acid at elevated temperature and pressure can rapidly decompose the oil matrix.17 Dry-ashing, which is carried out under normal air pressure in a muffle furnace, ensures that the organic matrix is completely mineralized and the total metal content is converted to simple water-soluble species.2,6 The wet-ashing technique, which is based on ignition of the organic material at high temperature and involves the use of strong acids and normal air pressure, also completely destroys the organic matrix.2 Ultrasonic energy, generated from an ultrasonic bath, is an alternative means of analyte extraction from a number of sample types.15,18 The volume of the water bath, sample position, percentage of detergent in the bath, and the bath temperature, are some of the most important variables that should be monitored.15 The objective of the study was to compare and evaluate the efficiencies of six analytical methods (xylene dilution, oil emulsification, microwave digestion, dry- and wet-ashing and ultrasonic extraction) used for the determination of trace metals in lubricating oils.
2. Experimental
2.1. Reagents and Standards
High purity doubly-deionized water, obtained using a Millipore Milli-Q deionizer (Millipore, Bedford, MA, USA), was used to prepare solutions. Analyzed Analytical Reagent (AAR) grade HNO3 (65 %), H2O2 (30 %), HCl (32 %), H2SO4 (98.08 %) and xylene (SMM Instruments (Pty) Ltd, South Africa) were used. A non-ionic surfactant (Triton X-100) obtained from Merck (Darmstadt, Germany) was used fordetergentformation. Aqueous working standards were prepared from 1000 mg kg-1 multielement stock solution, containing Ag, Ba, Cu, Mn and Ni in 5 % HNO3 (Teknolab AB, Kolbotn, Norway). Organo-metallic standard (Conostan S-21, Conoco Specialty Products Inc., Ponca City, OK, USA), with analyte concentration of 500 mg kg-1 in oil, was used for xylene dilution method. EnviroMAT, used oil certified reference material (CRM), HU-1 (SCP SCIENCE, Quebec, Canada) was used to check the accuracy of methods. Oil samples were collected from a local service station.
2.2. Instrumentation and Apparatus
Analysis of used lubricating oil for trace metals was carried out using a simultaneous Spectro Arcos ICP-OES with radial plasma viewing (Spectro Instruments, Kleve, Germany). The ICP-OES utilizes 32 linear CCD detectors in a Paschen-Runge mount. All ICP-OES-based analyses were monitored by Spectro Smart Analyser Vision Software (version 4.01) (Spectro Instruments, Kleve, Germany). The ICP-OES operational parameters employed are given in Table 1.
A MARS 5 microwave digestion system (CEM Corporation, USA) was employed for lubricating oil digestion. It was equipped with ESP-1500 plus pressure and RTP-300 plus temperature control systems. Teflon EasyPreps vessels, allowing a maximum decomposition pressure of 800 psi at 240 °C were used to digest the oils. An ultrasonic bath (Ultrasons, J.P. Selecta, Barcelona, Spain) was used for the ultrasound-assisted extraction and agitation of oil samples. Instrument operating parameters, like the intensity of the ultrasonic energy and temperature, were monitored by company default. Volumetric flasks of 100 mL were used as reaction vessels and placed at selected locations in the bath using burette supports.
2.3. Sample Preparation
2.3.1. Oil Dilution Procedure
A mass of 2.00 g used oil was accurately weighed and diluted to a final mass of 40.0 g with xylene. For calibration, appropriate quantities of organo-metallic standards were accurately weighed and made up to 2.00 g with base oil 75, after which diluted to 40.0 g with xylene. The CRM was also prepared in a similar manner as the working standards. In all analyzed solutions, the oil content was kept at 5 %, aiming to match the viscosities.
2.3.2. Oil Emulsification Procedure
Oil-in-water emulsions were prepared employing a specific sequence to guarantee their homogeneity and stability. An aliquot of 0.500 g used oil was placed in a 25.0 mL volumetric flask and 0.500 mL concentrated nitric acid was added aiming to dissolve some metals. The mixture was then agitated in an ultrasonic bath for 5 min followed by the addition of 1.00 g Triton X-100 to stabilize the oil-in-water emulsion. The resulting mixture was then shaken gently, after which doubly-deionized water was added, under continuous agitation, until a final mass of 10.0 g was obtained. The resulting mixture was finally shaken vigorously and agitated in an ultrasonic bath for 5 min to homogenize the contents. Calibration standards were prepared in a similar manner as the real samples, but using the required quantity of inorganic standard.
2.3.3. Microwave Digestion Procedure
An aliquot of used oil (0.200 g) was accurately weighed into an EasyPreps vessel liner. After addition of 4.00 mL nitric acid, the vessels were subjected to digestion based on the MARS 5 ramp-to-temperature heating programme of 1600 maximum power, 30 min ramp, 800 psi pressure, 200 °C maximum temperature and 10 min hold time. In each batch, six vessels were used for simultaneous digestion. The resulting digested material was then transferred to a pre-cleaned high density polypropylene (HDPP) vessel, and diluted to 20.0 g with doubly-deionized water.
2.3.4. Dry-ashing Procedure
A mass of 2.00 g used lubricating oil was accurately weighed in a porcelain crucible and heated on a hot plate until completely dried. The dried sample was then exposed to a two stage muffle furnace heating programme until completely ashed: 450 °C for 11 h (overnight) and 550 °C for 1.5 h. The resulting ash was then dissolved in 2.00 mL concentrated nitric acid and finally diluted to 40.0 g with Milli-Q water.
2.3.5. Wet-ashing Procedure
A mass of 2.00 g used lubricating oil was accurately weighed in a porcelain crucible. After additions of 2.00 mL sulphuric acid, the sample was subjected to heating on hot plate until completely dried. For Ba analysis, 1.50 mL of nitric acid was used in place of sulphuric acid, which was introduced dropwise to minimize sample splashing and foaming. The dried sample was then subjected to a two-stage muffle furnace heating programme until completely ashed: 450 °C for 2 h and 550 °C for another 2 h. The resulting ash was then dissolved in 1.50 mL concentrated HNO3, and finally diluted up to 40.0 g with Milli-Q water.
2.3.6. Ultrasound-assisted Extraction Procedure
A mass of 2.00 g used lubricating oil was accurately weighed and placed in a pre-cleaned 100 mL volumetric flask. An optimized amount of extractant solution (10 mL aqua regia) was then added and the resulting mixture irradiated at the optimum sonication time of 120 min to guarantee maximum sample irradiation, the volumetric flasks were kept stationary at selected positions in the bath and only four samples were used for simultaneous sonication. The resulting supernatant liquid was separated from the solid phase by centrifugation at 2500 rpm for 15 min after which diluted up to 40.0 g with Milli-Q water.
2.4. Factorial Optimization of Ultrasound-assisted Extraction Conditions
Ultrasonic energy, generated from an ultrasonic bath, was thoroughly studied for its capability to extract analytes of interest in used lubricating oil samples. A full factorial experimental design of 43 with 64 runs was proposed to study the effects of sonication time, extractant solution ratio and volume. The extracting solutions selected for this study were nitric and hydrochloric acids and hydrogen peroxide in different proportions, as shown in Table 2. Maximum analyte recovery was used as a response criterion and the three factors along with the four levels as control variables for the optimization scheme. Mass of oil (2 g), volume of water bath (2000 mL) and detergent content of the bath (2 %, v/v) were kept constant throughout the optimization scheme.
4. Results and Discussion
4.1. Optimization of Ultrasound-assisted Extraction
Sonication of used oil samples applying the full factorial design (43) produced a supernatant solution, which could easily be aspirated into the plasma. Analysis of the supernatant solution by ICP-OES, employing aqueous calibration standards, gave quantifiable data. These data were used to compute the recovery efficiency of each experimental condition, which finally used as criteria for deciding the optimum extraction conditions. Thus, sonicating 2.00 g used oil sample in 10.0 mL aqua regia for 120 min was found to be the optimum extraction condition for the elements studied.
4.1.1. Influence of Nitric Acid
Used oils sonicated with nitric acid alone appeared yellow in colour and gave poor recoveries for Ag (1.70-1.50 %) and Ba (15.9-79.9 %). However, it gave an enhanced extraction of Cu, Mn and Ni, with recoveries ranged from 52.3 to 106 %. Based on the calculated Pearson correlation coefficient (r), increasing the volume of nitric acid from 3 to 10 mL, while the sonication time was kept constant at 30 min, had a negative effect for the extraction of the elements studied, except for Ni. On the other hand, a simultaneous increase in the volume of nitric acid (3.00-10.0 mL) and sonication time (30-90 min) promoted a strong positive effect for all elements studied.
4.1.2. Influence of HNO3:H2O2 (2:1, v/v)
Used oil samples extracted with HNO3:H2O2 (2:1, v/v) appeared pale yellow in colour andhad an almost similar extraction effect when compared with using nitric acid alone. This extracting reagent yielded good recoveries of Cu, Mn and Ni ranged from 74.6 to 105 %; but poor recoveries of Agand Ba that ranged from 0.700 to 73.2 %. In general, increasing the sonication time, along with increasing the volume of this reagent, moderately enhanced the extraction efficiency of the metals studied.
4.1.3. Influence of HNO3:HCl (1:1, v/v)
Used oils extracted using HNO3:HCl (1:1) were very light yellow in colour and yielded good analyte recoveries of Cu, Mn, and Ni. The recoveries of Ag and Ba were still unsatisfactory but had improved when compared to the recoveries obtained using nitric acid alone and HNO3:H2O2 (2:1). At constant sonication time (30 min), increasing the volume of HNO3:HCl/1:1 from 3.00 to 10.0 mL caused a strong negative influence for the extraction efficency of Cu, Mn and Ni and a moderate negative influence for Ag and Ba. However, increasing the sonication time to 60 min together with an increase in the volume of HNO3:HCl/1:1, produced a strong positive association for Ag, Ba and Mn and moderate for Cu and Ni. Recoveries of Ag, Cu, Mn and Ni were generally observed to increase with increasing the volume of HNO3:HCl (1:1) from 3.00 to 10.0 mL.
4.1.4. Influence of Aqua Regia
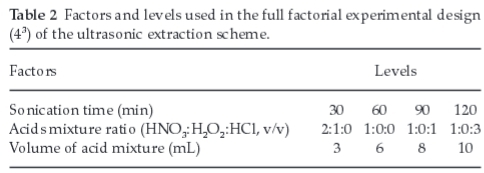
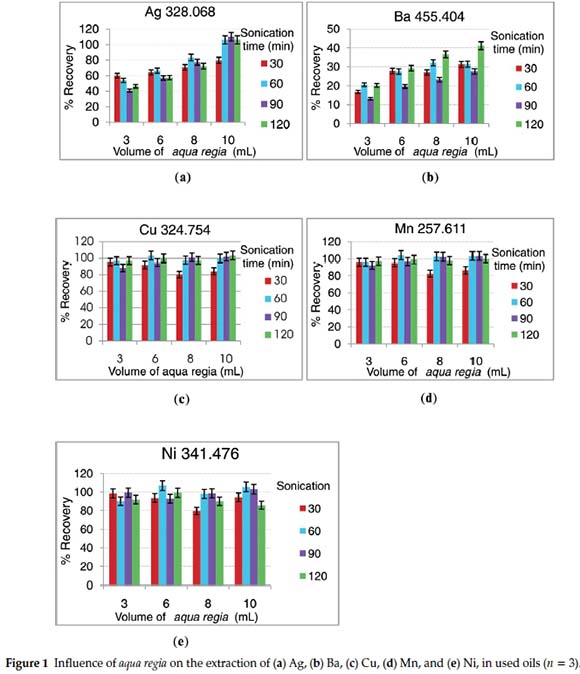
Used oil samples extracted using aqua regia (HNO3:HCl/1:3) gave a clear solution. As a strong oxidizing agent, aqua regia yielded good analyte recoveries of the elements studied, except for Ba (Fig. 1a-e). Further investigation revealed that under the same extraction conditions of 10.0 mL aqua regia and 120 min sonication time, the recovery of Ba increased as the spike level decreased from 5.95 to 2.33 μg g-1 (Fig. 2). The possible reason for this phenomenon lies on the limitation of the extracting agent (aqua regia) to isolate Ba analytes from the oil matrix as the spiked level exceeded about 0.450 μg g-1. Increasing the volume of aqua regia, while the sonication time was kept constant at 30 min, strongly promoted the recoveries of Ag and Ba. At the same time, it strongly suppressed the recoveries of Cu and Mn. When the sonication time was raised to 60 min; the recovery of Ag, Ba and Mn was highly increased with increasing the volume of aqua regia. Generally, a simultaneous increase in aqua regia volume and sonication time resulted in a better extraction efficiency for all the elements studied.
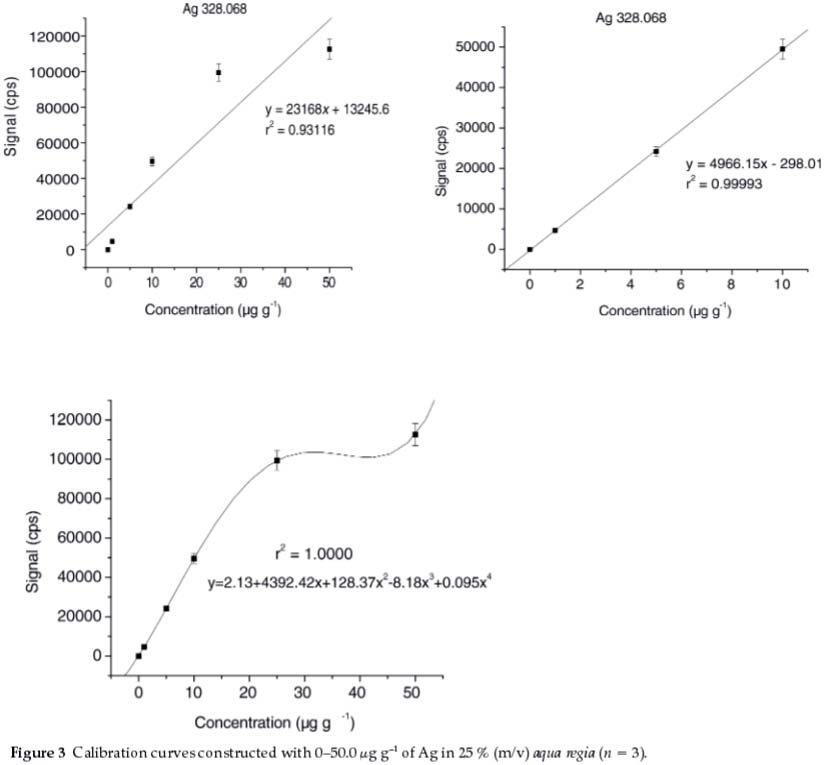
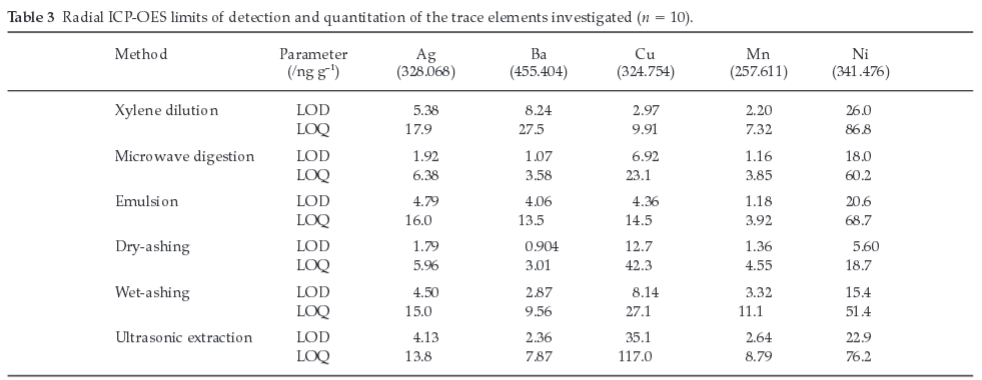
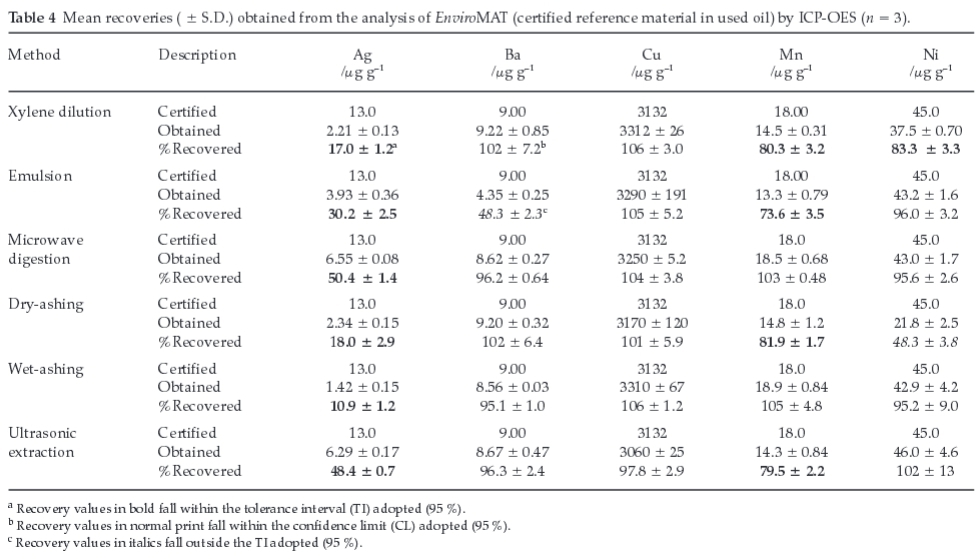
The recovery of Agwas shown to increase significantly with an increase in the volume of aqua regia from 3.00 to 10.0 mL (Fig. 1a). The increase in volume of aqua regia possibly has introduced more chloride ions into the solution, which might contribute to the formation of silver anionic complex (AgClx1-x) in solution, rather than a precipitate of AgCl. For this content, concentrations of Ag ranging from 0 to 10.0 μg g-1 in 25 % aqua regia (v/m) formed a clear solution and hence resulted in a good linearity (r2 = 0.99993), as depicted in Fig. 3. In contrast, calibration standards containing 25.0 and 50.0 μg g-1 of Agin 25 % aqua regia (v/m) were shown to form more precipitates of AgCl, and hence significantly deviated from the certified values (r2 = 0.93116). These results may imply that a reasonably low concentration of Ag is unlikely to form precipitate if kept in excess aqua regia.
4.1.5. Influence of Sonication Time
The effect of sonication time for the extraction of analytes in used lubricating oils was studied at 30, 60, 90 and 120 min. Increasing the sonication time was shown to increase the temperature of the bath (62 °C max), and hence the sample solution. When nitric acid was used as extractant, increasing the sonication time was generally observed to depress the recoveries of Ba and Ni; but was enhanced for Ag. Increasing the sonication time, during extraction with HNO3:H2O2 (2:1), generally increased the recoveries of Ag, Cu and Mn. When using low volumes of extracting reagents, usually 3.00 to 6.00 mL, increasing the sonication time was shown to have a slight influence on the extraction efficiency of the elements studied. Similarly, increasing the sonication time, employing 3.00 to 8.00 mL of aqua regia and HNO3:HCl (1:1) was observed to depress the recovery of Ag. A simultaneous increase in sonication time and concentration of the extracting reagents was generally found to enhance the extracting efficiency of the elements studied. This was critical, particularly when aqua regia and HNO3:HCl (1:1) were used as extracting agents.
4.2. Figures of Merit and Validation
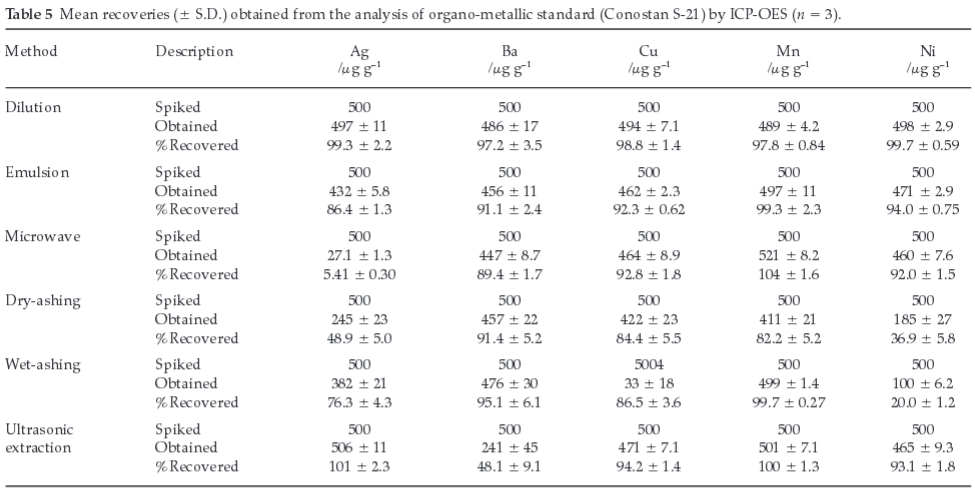
The limits of detection, LOD (3 Sb/m, where Sbis the standard deviation of 10 replicates of blank solutions and m the sensitivity) and limits of quantitation, LOQ (10 Sb/m) were computed for each element and they were in the low ng g-1 range (Table 3), which will easily permit the determination of trace levels of wear metals and trends to be observed. Microwave digestion and dry-ashing methods offered much better LOD and LOQ when compared with the other methods investigated.
A certified reference material (CRM) in used oil and organo--metallic standard were analyzed by ICP-OES. The quantitative data obtained from both quality control (QC) samples was used to compute the accuracy of each studied method. The recoveries obtained from both QC samples were comparable for most of the studied elements, as shown in Tables 4 and 5. Applying the Student's t-test to these data indicated good agreement between the certified and measured values at the 95 % confidence level, for most of the elements studied.
Each of the six analytical methods investigated were validated based on the criteria illustrated in Table 6 and the method of choice for the determination of Mn was microwave digestion; for Cu, xylene dilution; for Ba, wet-ashing; for Ni, oil emulsification and for Ag ultrasonic extraction. The methods investigated were further evaluated in terms of suitability for routine analysis, sample preparation time and cost (approximate cost of reagents and instrument running costs). Based on these evaluations, an ultrasonic extraction has a clear advantage in terms of accuracy, cost of sample preparation, simplicity, safety and suitability for routine analysis.
4.3. Evaluation of Sample Preparation Methods
4.3.1. Xylene Dilution Method
The dilution method required the use of organo-metallic standards for calibration, which were expensive when compared to inorganic standards. This technique did not minimize the organic load of the oil and hence plasma stability was a concern and which demanded careful optimization of the gas flow rates. The choice of special sample tubes that can withstand organic solvents also contributed to an additional cost of this method. On the other hand, the dilution method offered minimal sample contamination; good accuracy (97.2-99.7 %); good precision (0.591-2.85 % RSD), except for Ba (7.03 %); good LOQs (7.32-86.8 ng g-1) and good linearity with r2ranging from 0.9996 to 0.9998. It was also easier to handle, cheaper and rapid and hence it can be applied for routine analysis.
4.3.2. Oil Emulsification Method
The oil-in-water emulsion required sequential sample preparation and small sample size so as to form a stable and homogenous emulsion. It was often difficult to form stable and homogenous emulsions with matched viscosity, particularly for viscous oils. In contrast, it was cheap and fast, allowed aqueous standards for calibration and gave good linearity with r2ranging from 0.9998 to 1.000. This method yielded good analyte recoveries (86.4-99.3 %) for all the elements studied when additions of Conostan standards were used. However, it yielded unsatisfactory recoveries for Ag and Ba in the CRM. This variation in accuracy might have occurred due to the low level of these elements in the CRM. The precision of results determined for this method were fairly good, with %RSDs ranging from 1.53 to 4.99 %. The calculated LOQs were in the range of 3.92 to 68.7 ngg-1. These LOQ values imply that the method could be used for the determination of trace levels of wear metals in lubricating oils.
4.3.3. Microwave-assisted Acid Digestion Method
The pressurized microwave digestion gave complete digestion of oil samples, which could easily be aspirated into the ICP-discharge. The closed microwave digestion technique offered minimal sample contamination, good precision (0.467-3.63 % RSD) and good accuracy (95.6-104 %), except for Ag (50.4 %). The LODs for this method were low, ranging from 1.07 to 18.0 ng g-1. In contrast, the initial and running costs of the microwave apparatus were high, which could limit its use for routine analysis. The limited organic sample size recommended for digestion was also a major drawback as it may not have resulted in a representative sample. In addition, this method is moderately unsafe due to the probability of vessel explosion caused by faults such as: vessel leaks, sample size, sample matrix and operator's error.
4.3.4. Dry-ashing Method
The dry-ashing method completely mineralized the oil matrix and resulted in a clear solution, which could be easily aspirated into the plasma. It was simple to use and control all steps of the procedure. Accuracy of the method was also moderately good (81.9-102 %), with the exception of Ag (48.9 %) and Ni (48.3 %). However, the precision of these results was poor, ranging from 5.80 to 16.2 % RSD, except for Mn (2.06 %). The LODs obtained by this method were low, ranging from 0.904 to 12.7 ng g-1. On the other hand, sample preparation time was rather long (about 17 h), which limits its applicability for routine analysis.
This long and open-air sample heating can obviously cause sample contamination, which is a major concern in trace metal analysis. Loss of some volatile analytes during the intense heating and muffling was a major concern. Consequently, a significant loss of Ni for both types of additions occurred and this resulted in reduction of Ni recovery from 49.8 to 7.67 % as the spiked level increased from 1.18 to 10.0 μg g-1. Hence the dry-ashing technique was found to be inefficient for the determination of Ni.
4.3.5. Wet-ashing Method
The wet-ashing method completely destroyed the oil matrix and resulted in a clear solution. When compared to dry-ashing, the wet-ashing method offered a relatively short sample preparation time, taking 6 h (2 h heating to dryness and 4 h muffling). Sample heating and muffling employed in both dry and wet-ashing procedures were relatively inexpensive when compared to the microwave digestion procedure. Hence, the wet-ashing approach can be used for routine analysis of oils, as an alternative method to microwave digestion. This technique generated lower LODs (2.87-15.4 ng g-1), good accuracy (95.1-106 %), except for Ag (76.3 %) and good precision (1.10-5.62 % RSd), except for Ni (9.47 %). Like dry-ashing, wet-ashing also yielded a poor recovery of Ni (38.7 %) when only nitric acid was used as oxidizing agent. Nonetheless, wet-ashing, employed concentrated sulphuric acid as oxidant, followed by nitric acid dissolution, minimized Ni losses and offered better recovery that fell within the confidence limit adopted (95 %). It is possible that Ni was retained as non-volatile NiS, which is sparingly soluble in nitric acid. This phenomenon is in agreement with the finding of Aucelio et al.19 Another interesting observation in the analysis of the QC samples, containing around 1μg g-1 of Ni, ashed using 2.00 mL of sulphuric acid, yielded a better recovery for Ni (95.2 %). However, increasing the concentration of Ni to 5.79 μg-1, using the same ashing conditions, resulted in an abrupt loss of Ni (loss of 80 %). This phenomenon may indicate the need for optimizing the quantity of sulphuric acid that can efficiently retain a given concentration of Ni. Sample contamination, formation of AgCl and BaSO4 precipitate when hydrochloric and sulphuric acids were used, and loss of Ni when nitric acid was used as oxidizing agent, sample loss due to foaming and sample adsorption into the porcelain crucibles were some major observed limitations for the wet-ashing technique.
4.3.6. Ultrasound-assisted Extraction
The ultrasound extraction approach, employing extractant reagents, was effective for the extraction of metals from used lubricating oils. An ultrasonic bath is inexpensive and simple to operate. The reaction vessels (volumetric flasks) employed were also inexpensive and available in any laboratory. The extraction procedure is safe and simple to handle, and required 2 h. This technique offered good accuracy (96.3-102 %), good precision (2.26-2.97 % RSD), except for Ni (12.6 %) and low LODs (2.36-35.1 ng g-1). However, the uneven distribution of the ultrasonic energy within the bath, which created cavitational variations, was found to be a major limitation of this technique. Consequently, only four samples were used in each batch to guarantee maximum ultrasonic irradiations. Even so, the ultrasonic extraction approach can be applied for routine analysis of oils for its low cost, fairly short sample preparation time, simple procedure and fairly good accuracy.
5. Conclusions
The recoveries obtained by ultrasonic-assisted extraction technique were quantitative, presenting values better than 93 %.
Such values denote that the ultrasonic extraction procedure, proposed in this work, is efficient in destroying the organic matrix, allowing for the releasing of metals. It was demonstrated that the most important interaction between variables occurred when a simultaneous increase in sonication time (30-120 min) and volume of the extracting reagent (3.00-10.0 mL) was applied. This was critical, particularly when HNO3:HCl (1:1, v/v) and HNO3:HCl (1:3, v/v), were used as extracting reagents. Furthermore, aqua regia was found to be the most powerful and universal extracting reagent for all metals studied.
The results obtained from the analysis of additions of both organo-metallic standard (Conostan S-21) and CRM by ICP-OES, permitted validation of each method studied with respect to its accuracy, precision, linearity, LOD and LOQ. Based on these evaluations, the choice of method for the determination of Mn in used oils was microwave digestion, for Ba, wet-ashing; for Cu, xylene dilution; for Ni, detergent emulsion and for Ag, ultrasonic extraction. Further evaluation of the methods in terms of suitability for routine analysis, sample preparation time and cost (cost of reagents and instrumental running costs) indicated the technique of ultrasound extraction has a clear advantage in terms of accuracy, cost of sample preparation, simplicity for use, safety and suitability for routine analysis. In general, evaluation of the six analytical methods investigated (xylene dilution, detergent emulsion, microwave digestion, dry-ashing, wet-ashing and ultrasonic extraction) for the determination of trace elements in used lubricating oils showed significant differences when statistically compared using the analysis of variance (ANOVA) at the 95 % confidence level.
Acknowledgements
The authors gratefully acknowledge Tshwane University of Technology and NRF for funding this project.
References
1 R.O. Aucélio, G.P. Brandáo, R.C. Campos, C. Duyck, P. Grinberg, N. Miekeley and C.L.P Silveira, The determination of trace metals in lubricating oils by atomic spectrometry, Spectrochim. Acta Part B, 2007,62, 952-961. [ Links ]
2 T. Kuokkanen, S. Lahdelma, K. Roppola, P. Vahaoja and I. Valimaki, Wear metal analysis of oils, Crit. Rev. Anal. Chem., 2008, 38, 67-83. [ Links ]
3 P.J.J.G. Marais and M.J. Orren, Application ofan Argon-cooled Inductively Coupled Plasma to the Analysis of Metals in Lubricating Oils, MSc dissertation, University of Cape Town, Cape Town, South Africa, 1987. [ Links ]
4 B. Jeremic and I. Macuzic, Development of mobile device for oil analysis, Tribol. Indust, 2004,26, 27-31. [ Links ]
5 V.R.A. Filho, and J.A.G. Neto, Evaluation of lubricating oil preparation procedures for the determination of Al, Ba, Mo, Si and V by high-resolution continuum source FAAS, Anal. Sci., 2009,25,95-100. [ Links ]
6 I.M. Goncalves, A.M. Gonzaález and M. Murillo, Determination of metals in used lubricating oils by AAS using emulsified samples, Talanta 1998,47, 1033-1042. [ Links ]
7 R.Q. Aucelio, C.L.P da Silveira and R.M. Souza, Evaluation and validation of instrumental procedures for the determination of nickel and vanadium in fuel oils, Anal. Sci. 2004, 20, 351-355. [ Links ]
8 P. Yaroshchyk, R.J.S. Morrison, D. Body, and B.L. Chadwick, Quantitative determination of wear metals in engine oils using laser-induced breakdown spectroscopy: a comparison between liquid jets and static liquids, Spectrochim. Acta, Part B, 2005, 60, 986-992. [ Links ]
9 J. Chirinos, A. Fernandez and J. Franquiz, Multi-element optimization of the operating parameters for inductively coupled plasma atomic emission spectrometry with a charge injection device detector for the analysis of samples dissolved in organic solvents, J. Anal. At. Spectrom, 1998,13, 995-1000. [ Links ]
10 R. Carpenter and L. Ebdon, A comparison of inductively coupled plasma torch - sample introduction configurations using simplex optimization, J. Anal. At. Spectrom, 1986,1, 265-268. [ Links ]
11 R.M. de Souza, L.G. Leocádio, and C.L.P. da Silveira, ICP OES simultaneous determination of Ca, Cu, Fe, Mg, Mn, Na, and P in biodiesel by axial and radial inductively coupled plasma-optical emission spectrometry, Anal. Lett, 2008 41, 1615-1622. [ Links ]
12 E. de Oliveira, Sample preparation for atomic spectroscopy: evolution and future trends, J. Braz. Chem. Soc., 2003,14, 174-182. [ Links ]
13 L.M. Costa, S.L.C. Ferreira, J.A. Nóbrega and A.R.A. Nogueira, Use of factorial design for optimization of microwave-assisted digestion of lubricating oil J. Braz. Chem. Soc., 2005,16, 1269-1274. [ Links ]
14 R F.W.S. Ana, A.R. Cassella, R.J. Cassella and R.E. Santelli, Optimization of an open focused microwave oven digestion procedure for determination of metals in diesel oil by inductively coupled plasma optical emission spectrometry, J. Hazard. Mater., 2007,149, 67-74. [ Links ]
15 J.L. Capelo, C. Maduro and C. Vilhena, Discussion of parameters associated with the ultrasonic solid-liquid extraction for elemental analysis (total content) by electrothermal atomic absorption spectrometry. An overview, J. Ultrason. Sonochem., 2005, 12, 225-232. [ Links ]
16 K. Jankowski, A. Jerzak, A. Sernicka-Poluchowicz and L. Synoradzki, Multielement determination of major elements in polymer additives by microwave induced plasma atomic emission spectrometry after microwave digestion, J. Anal. Chim. Acta, 2001, 440, 215-221. [ Links ]
17 A.M. Essling, D.R Huff, E.A. Huff, I.M. Fox and D.G. Graczyk, Preparation of waste oil for analysis to determine hazardous metals, ANL/ACL-95/ I, Department of Commerce, National Technical Information Service, USA, 1995, pp. 1-28. [ Links ]
18 R.A.A. Munoz, P.V. Oliveira, and L. Angnes, Combination of ultrasonic extraction and stripping analysis: An effective and reliable way for the determination of Cu and Pb in lubricating oils, Talanta, 2006, 68, 850-856. [ Links ]
19 R.Q. Aucélio, G.P. Brandáo, R.C. Campos, C. Duyck, P. Grinberg, N. Miekeley and C.L.P Silveira, The determination of trace elements in crude oil and its heavy fractions by atomic spectrometry, Spectrochim. Acta, Part B, 2007a,62, 939-951. [ Links ]
Received 14 November 2014
Revised 8 April 2015
Accepted 10 April 2015
Dedicated to the late Prof. P.J.J.G. Marais
* To whom correspondence should be addressed: E-mail: mccrindleri@tut.ac.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}