










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Chemistry
versão On-line ISSN 1996-840X
versão impressa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.68 Durban 2015
http://dx.doi.org/10.17159/0379-4350/2015/v68a5
RESEARCH ARTICLE
http://dx.doi.org/10.17159 /0379-4350/2015/v68a5
Synthesis and NMR elucidation of novel octa-amino acid resorcin[4]arenes derivatives
Iman ElidrisiI; Pralav V. BhattII; Thavendran GovenderII; Hendrik G. KrugerII; Glenn E. M. MaguireI, *
ISchool of Chemistry and Physics, University of KwaZulu-Natal, Westville Campus, Private Bag X54001, Durban, 4000, South Africa
IISchool of Pharmacology, University of KwaZulu-Natal, Westville Campus, Private Bag X54001, Durban, 4000, South Africa
ABSTRACT
The synthesis of nine novel protected amino acid cavitands is reported. All have four pendant M-undecyl chains and 'headgroups' connected by a two-carbon spacer at eight positions on the aromatic rings. The amino acids employed are glycine, alanine, phenylalanine, leucine, proline, tryptophan, serine, glutamine and lysine. The structures of the compounds were elucidated using one and two-dimensional NMR techniques which verified that all octa-substituted cavitands have symmetrical C2vconformation at room temperature. These compounds have potential synthetic ion channel applications.
Keywords: Octa-amino acid resorcin[4]arenes, 1H-NMR, COSY, HSQC, C4v symmetry, C2v symmetry.
1. Introduction
Resorcin[4]arenes are well-known macrocyclic oligomers formed when resorcinol condenses with aliphatic or aromatic aldehydes under acidic conditions.1 The reaction with formaldehyde is excluded from this 'family' as it often forms linear polymers. Even though it is possible to form resorcinarenes with formalde-hyde,2 the use of aliphatic aldehydes resulting in side chains or 'feet' is preferred for potential synthetic ion channels.3 These macrocyclic compounds are known to possess hydrophilic (upper rim) and hydrophobic (lower rim) regions and a cavity, that can accommodate small organic molecules.4
Resorcin[4]arenes are not planar and can adopt five possible conformational arrangements: the C4vsymmetrical 'crown' conformation, C2vsymmetrical 'boat' conformation, C2hsymmetrical 'chair' conformation, Cssymmetrical 'diamond' conformation, and S4symmetrical 'saddle' conformation as illustrated in Fig. 1.4b
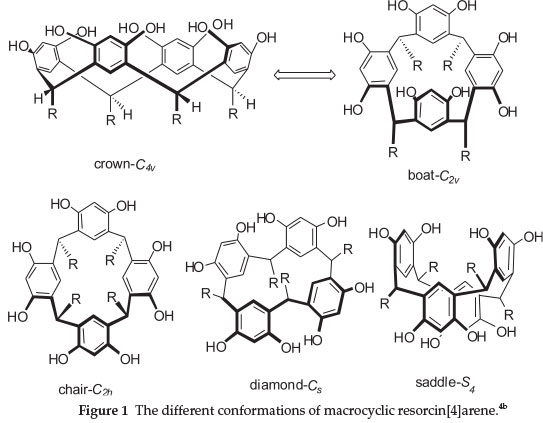
The presence of two electron-releasing hydroxyl groups on the aromatic rings especially at the 'ortho' position makes compounds of this family a convenient platform for the design and synthesis of various supramolecular structures. To obtain these architectures, various methods have been developed for selective chemical modifications of the resorcin[4]arenes.4b,5 Functionalization of the resorcin[4]arene platform with amino acid moieties could create structural features that provide valuable insight into factors governing biologically relevant host-guest chemistry.6 These types of compounds also have found applications as synthetic ion channels.3,7.
This study demonstrates an effort to functionalize resorcin[4] arene with amino acid residues at the upper rim. Very few examples have been reported where 'flexible' resorcin[4]arenes have been modified with amino acids. An example employing L-proline via Mannich reactions has been reported by several researchers.8 Botta et al. using a different approach have modified resorcin[4]arenes at the lower rim with several amino acids for chiral recognition.9
The synthesis of our target compounds began by utilizing dodecanal, as the alkyl aldehyde component for the condensation reaction with resorcinol, to produce the resorcin[4]arene, 1 in good yield as reported in literature.1b,10 Acylations and alkylations of hydroxyl groups have produced cavitands, carcerands, hemicarcerands, velcrands,4a molecular capsules,11 receptors and sensors for biologically-active compounds,12 and metal ion extraction agents.13
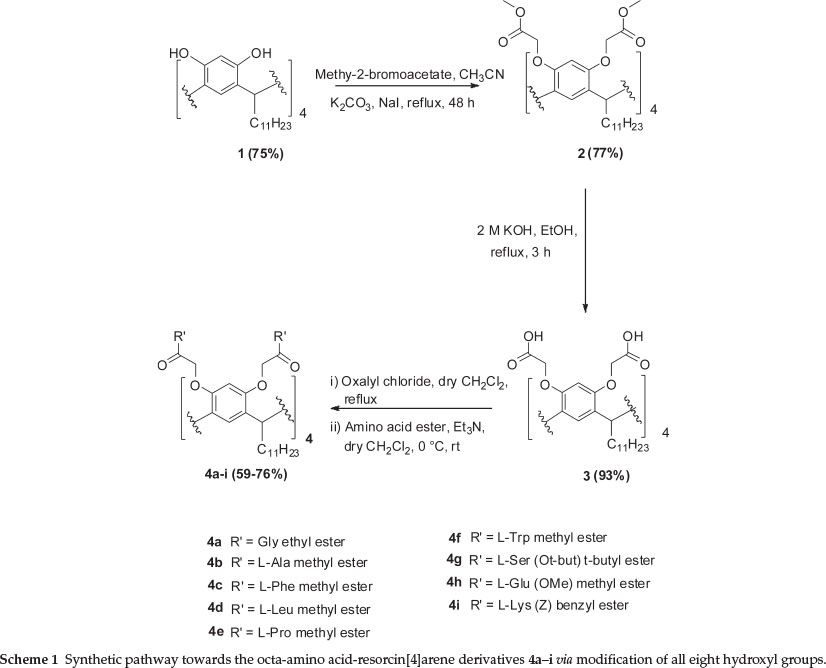
The synthetic route towards novel compounds 4a-i (Scheme 1), involves alkylation of compound 1 with methyl-2-bromoacetate in dry acetonitrile in the presence of potassium carbonate and a catalytic amount of sodium iodide. The reaction occurs at elevated temperature for 48 h and after workup and recrystal-lization pure 2 was obtained in 77 % yield.13a14
2. Results and Discussion
These compounds may find application as synthetic ion channels, but testing of the compounds reported herein for that, falls outside the scope of the current NMR investigation.
The structures of these compounds were established on the basis of one- and two-dimensional NMR experiments. A discussion of the complete elucidation of compound 4a is presented, followed with a short discussion of 2D results for 4b. Elucidation of the remainder of the compounds is presented in the online supplement. A summary of the NMR data are presented in Tables 1-3.
Octa-acid resorcin[4]arene 3 was transformed into the octa-acyl chloride upon treatment with oxalyl chloride in dry CH2Cl2 (Scheme 1). Since the acyl chloride is very unstable and undergoes rapid hydrolysis if moisture is present, it was used in the next step without further purification and characterization. This acyl chloride was reacted with nine equivalents of each amino acid, to the get the novel derivatives.15 A number of the amino acids required side chain protection (e.g. L-glutamic acid, L-serine, and L-lysine). The crude materials obtained were purified on silica gel chromatography, using 3 % methanol in chloroform. The octa-amino acid resorcin[4]arene derivatives 4a-i were obtained in good yields (59-76 %).
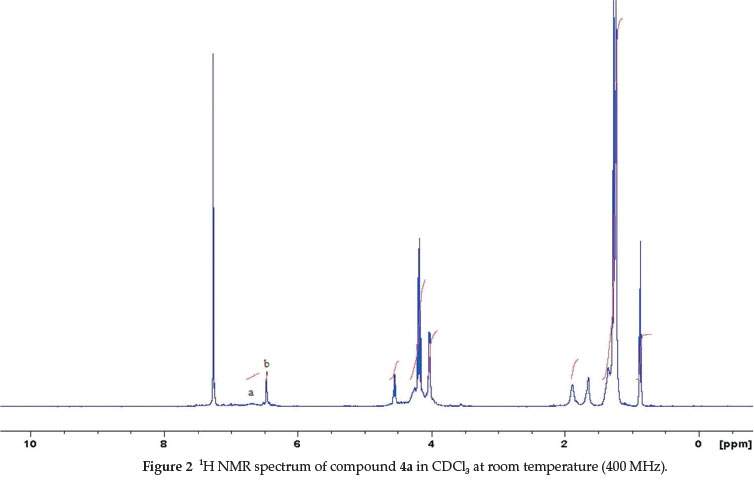
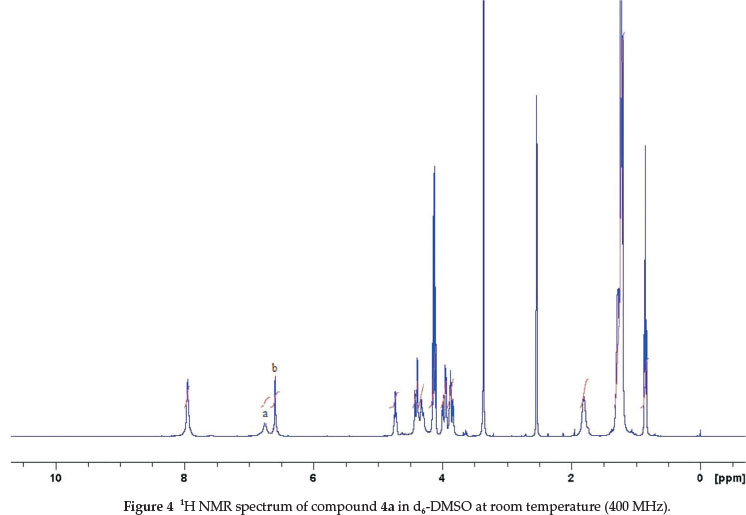
The 1H NMR spectra of these derivatives (4a-i) in CDCl3 at room temperature showed a considerable broadening of the various signals. The relatively broad signals from these compounds are a result of the multiple conformations possible for the cavitand bowl. On the NMR timescale, they have a slow rate of interconversion. Boat conformations convert to a symmetrical crown conformation and vice versala,10a,16. A similar observation was made when the spectra of these molecules were taken in polar organic solvents at room temperature (d6-acetone and d6-DMSO). Figures 2, 3 and 4 illustrate these observations for compound 4a in different solvents. The 1H NMR spectra of this derivative show relatively broad signals corresponding to the aromatic protons (assigned a and b).
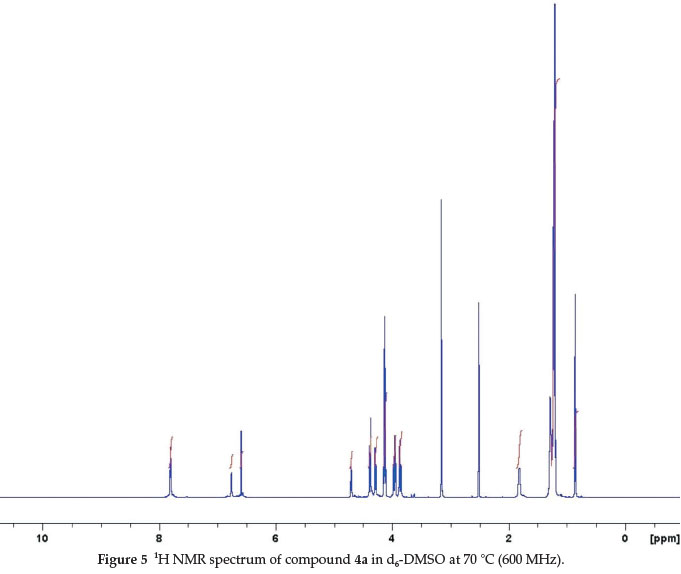
In an attempt to confirm that the broadening in the 1H NMR spectra is the result of these conformational changes, 4a in d6-DMSO was heated in the NMR spectrometer (600 MHz). By increasing the temperature stepwise, the various signals began to sharpen. At 70 °C, only the signals of the crown conformation are visible in the spectrum. 1H NMR spectrum of this compound shows sharp single peaks corresponding to the aromatic protons (assigned at 6.73 ppm and 6.57 ppm) (Fig. 5).
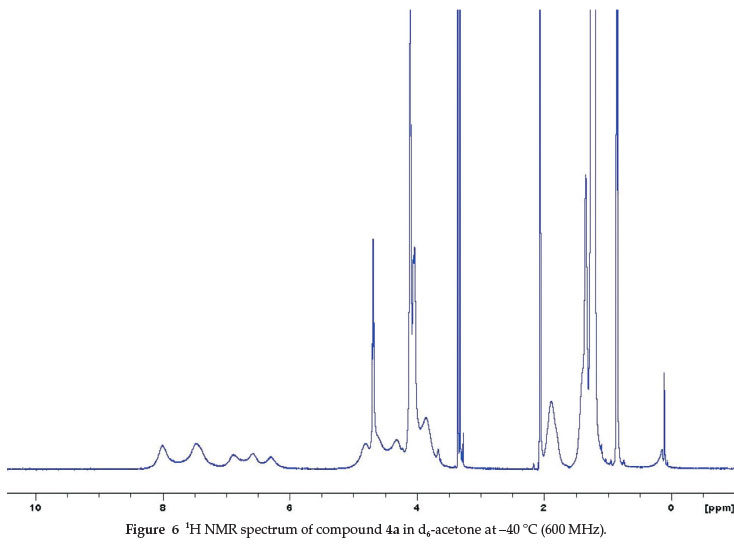
To further establish the conformational behaviour for these compounds (4a-i), a low temperature 1H NMR experiment was recorded for compound 4a in d6-acetone at 600 MHz from 0 °C to -60 °C. The most notable change in the 1H NMR spectrum occurred for the signals of the aromatic protons (H3 and H4, Fig. 3). As the solution cooled down to -40 °C, the signals for H3 and H4 broadened and separated into four signals (6.30 ppm and 6.57 ppm for H, and 6.89 ppm and 7.47 ppm for H3) (Fig. 6).
Figure 6 illustrates the splitting of the aromatic protons signals into four broad singlets at low temperature, indicating the flattened conformations corresponded to a C2vsymmetry for 4a, in which the aromatic rings lie spatially in pairs. As anticipated, the 1H NMR spectrum in Fig. 3 also displayed some changes in the non-aromatics regions for this compound.
To discuss the proton (1H) NMR spectrum of compound 4a in DMSO-d6 at 70 °C reference will be made to Fig. 7, which shows the numbering of the protons present in this molecule. According to this expanded structure, the various resonances present in the 1H NMR spectra of the macrocycles 4b-i were assigned.
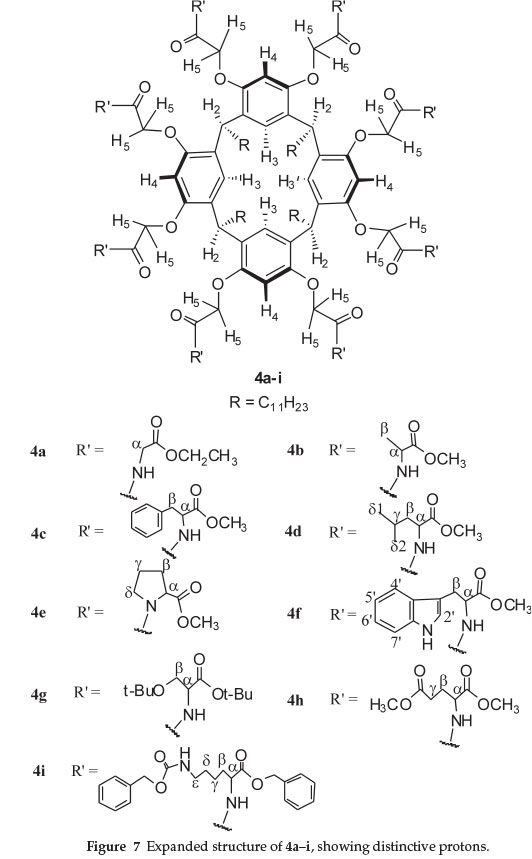
The 1H NMR spectrum for compound 4a in d6-DMSO at 70 °C demonstrates signals characteristic for the glycine ethyl ester and the resorcin[4]arene scaffold. The signal related to the methylene group of the ethyl ester at 4.12 ppm appears as a quartet, integrating to 16. The signal associated with the methyl group of this ester at 1.20 ppm is a triplet, integrating to 24. The signal related to the α-protons (Fig. 7) appears as two pairs of doublets at 3.96 ppm and 3.85 ppm, each of these integrates to eight. The signal for the amide NH protons for this derivative appears as a triplet at 7.72 ppm, integrating to eight.
The signal for the methylene protons (H5) of the OCH2CO groups appears as two doublets at 4.36 ppm and at 4.27 ppm, each of these signals integrates to eight. This splitting could be attributed to the presence of two glycine residues on each aromatic ring. The signals related to the protons of aromatic rings (H3 and H4) appear as two singlets at 6.73 ppm for H3 protons and at 6.57 ppm for H4 protons, each of these integrates to four. The signal associated with the methine protons (H2)at 4.69 ppm is a triplet, integrating to four. The signals related to the undecyl 'feet' (R) have resolved into three signals: a quartet at 1.81 ppm, integrating to eight, multiplets at 1.22-1.27 ppm, integrating to 72, and a triplet at 0.84 ppm, integrating to 12.
The IR spectrum for 4a shows the characteristic appearance of the amide NH stretching peak at 3414 cm-1 whereas the carbonyl peaks at 1757 and 1731 cm-1 corresponding to the ester and amide carbonyl stretching frequencies, respectively. Mass spectrometry (MS) (using ESI-TOF methods) additionally gave a molecular ion m/z signal of 2273.2926, which matches the expected mass for 4a of 2273.2942.
Compound 4b was synthesized in 73 % yield by reacting L-alanine methyl ester with the octa-acid resorcin[4]arene 3. The 1H NMR data for compounds 4a-c are summarized in Table 1.
Subsequent COSY and HSQC NMR analysis confirmed the presence of the target compounds, showing the expected couplings between the various protons. Figure 7 displays 1H-1H COSY couplings for compound 4b, as an example of a two-dimensional NMR experiment at 70 °C (600 MHz.)
Analysis of the COSY spectrum shows (Fig. 8) that the α-protons (Ala-CH-) at 4.35 ppm are coupled to the amide NH protons at 7.82 ppm and 7.80 ppm as well as the β-protons (Ala-CH3)at 1.34 ppm. The methine protons (H2) at 4.67 ppm which bridge the aromatic moieties, are coupled to the methylene protons of the 'feet' (-CH2-) at 1.83 ppm. The methylene protons at 1.83 ppm are coupled to the protons of the 'feet' at 1.20-1.28 ppm. The terminal methyl groups of the 'feet' at 0.84 ppm are coupled to the methylene groups at 1.20-1.28 ppm.
Compound 4d was synthesized in 72 % yield by reacting L-leucine methyl ester with the octa-acid resorcin[4]arene 3. Reaction of the octa-acyl chloride resorcin[4]arene 3 with L-proline methyl ester afforded compound 4e in 69 % yield. The octa-acyl chloride resorcin[4]arene 3 reacted with L-proline methyl ester afforded compound 4e in 69 % yield. Compound 4f was synthesized in 64 % yield by reacting L-tryptophan methyl ester with the octa-acid resorcin[4]arene 3. The 1H NMR data for compounds 4d-f are summarized in Table 2.
Subsequent COSY and HSQC NMR analysis confirmed the presence of the target compounds, showing the expected couplings between the various protons.
Reaction of O-t-butyl-L-serine t-butyl ester with the acyl chloride resorcin[4]arene gave compound 4g in 59 % yield. Compound 4h was synthesized in 60 % yield by reacting L-glutamic acid dimethyl ester with the octa-acid resorcin[4]arene 3. Compound 4i was isolated in 67 % yield by reacting Νε-Cbz-L-lysine benzyl ester with the octa-acid resorcin[4]arene 3. The 1H NMR data for compounds 4g-i are summarized in Table 3.
Subsequent COSY and HSQC NMR analysis confirmed the presence of the target compounds, showing the expected couplings between the various protons.
With reference to Fig. 7, the 1H NMR data (Tables 1, 2 and 3) for these compounds clearly demonstrated that: all compounds had characteristic appearance of the amide NH protons, which appear at a lower frequency indicating the involvement of these protons in intermolecular hydrogen bonding with the water molecules present in the NMR solvent (DMSO), except compound 4e.17 The signal for the diastereotopic methylene protons (H5, Fig. 6) appears as a pair of quartets in the 1H NMR spectra for compounds 4b, 4c, 4d, 4f, 4g, 4h, and 4i. This splitting can be attributed to the presence of two chiral amino acid units on each aromatic ring15a,15b. In compound 4a where there is no chiral centre, this signal appears as a pair of doublets since the protons are enantiotopic.
The signals for the aromatic protons (H3 and H4) appear as two singlets for all compounds. This indicates the symmetric positions of these protons in a crown (C4v) conformation at elevated temperature. Therefore, at high temperature, the rate of conformational interchange is high on the NMR timescale and only the signals associated with the crown conformation are observed10a.
The signal related to the methine protons (H2) which bridge the aromatic moieties, appears as a triplet in 1H NMR spectra for these compounds. The signals associated with the undecyl 'feet' remain essentially unchanged in terms of multiplicity and integration. However, these protons have experienced very small changes in terms of chemical shift.
3. Conclusion
A series of new resorcin[4]arenes appended with amino acid moieties at the upper rim (4a-i) were synthesized in good yields (59-76 %). The key step for this synthesis is the amide bond formation between the amine functional group of amino acid units and carboxylic acid group of the octa-acid resorcinarene, 3. The structure of these octa-substituted resorcinarene derivatives was established on the basis of one and two-dimensional NMR experiments, and confirmed by IR and MS spectra. The 1H NMR data obtained for the protons related to the aromatic rings of resorcin[4]arene scaffold (H3 and H4, Fig. 6) verified that these derivatives adopted stable crown conformations (C4vsymmetry) at high temperature. Low temperature NMR experiments confirmed that a boat conformation predominates for these compounds, which is expected for resorcinarenes with eight bulky substituents. These structures are very similar to previously reported ion channel molecules; we presume that they will be capable of ion translocation activity.
Experimental
Starting materials obtained from commercial suppliers were used without further purification unless otherwise stated. Air- or moisture-sensitive reactions were performed using oven-dried glassware under an inert atmosphere of dry nitrogen. Tetrahy-drofuran (THF) was distilled from sodium benzophenoneketyl and dichloromethane was distilled from calcium hydride. Dimethyl sulfoxide (DMSO) and dimethylformamide (DMF) were stored over 4 Ẳ molecular sieves prior to use. Thin layer chromatography (TLC) was performed on aluminium-backed, pre-coated silica gel plates (Merck, silica gel 60 F254, 20 cmx 20 cm). Mobile phases are reported as volume ratios or volume per cent. Compounds were visualized using UV light, p-anisaldehyde, or iodine stains. Column chromatography was performed on silica gel 60 (Merck, particle size 0.040-0.063 mm). Eluting solvents are reported as volume ratios or volume per cent. Melting points were recorded and are uncorrected. 1H NMR spectra were recorded on a 400 MHz Bruker AVANCE III or 600 MHz BrukerUltrashield spectrometer, the chemical shifts were referenced to the solvent peak, namely δ = 7.24 ppm for CDCl3, δ = 2.50 ppm for (CD3)2SO and δ = 2.05 ppm for (CD3)2CO at ambient temperature. The 1H NMR spectra were recorded at a transmitter frequency of 600.1 MHz (spectral width, 12335.5 Hz; acquisition time, 1.328 s; 90 ° pulse width, 15 μs; scans, 16; relaxation delay, 1.0 s) for the Bruker AVANCE III 600 instrument while the 1H NMR spectra were recorded at a transmitter frequency of 400.2 MHz (spectral width, 8223.7 Hz; acquisition time, 3.98 s; 90 ° pulse width, 10 μs; scans, 16; relaxation delay, 1.0 s) for the Brucker AVANCE III 400 instrument. The 13C NMR spectra were recorded at 150.9 MHz (spectral width, 36057.7 Hz; acquisition time, 0.908 s; 0.908 s, 90 ° pulse width, 9.00 μs; scans, 4800; relaxation delay, 2.00 s) for the Bruker AVANCE III 600 instrument while the 13C NMR spectra were recorded at 100.6 MHz (spectral width, 24038.5 Hz; acquisition time, 1.363 s; 90 ° pulse width, 8.40 μs, scans, 3200; relaxation delay, 2.00 s) for the Bruker AVANCE III 400 instrument.
The 2D experimental data parameters obtained on the Bruker AVANCE III 400 were as follows: 90 ° pulse width, 10 μs for all spectra, spectral width for 1H, 3731.3, 3521.1, 3401.3, 3676.4, 3125.0 and 4065.0, 4065.0, 3731.3, 3546.1 Hz for 4a, 4b, 4c, 4d, 4e, 4f, 4g, 4h and 4i, respectively, (COSY and HSQC) spectral width for 13C, 166670.4 Hz (HSQC) for 4a-i, number of data points per spectrum, 2048 (COSY), 1024 (HSQC) for compounds 4a-i; number of time incremental spectra 128 (COSY), 256 (HSQC) for compounds 4a-i; relaxation delays for compounds for compounds 4a, 4f, 4g, 4h and 4i was 1.4s and 4b, 4c, 4d and 4e was 1.3 s for COSY experiments while the relaxation delay for HSQC experiments had 1.4 s for 4a-i, respectively. Data are reported as positions in parts per million (δ in ppm), multiplicity (s = singlet, d = doublet, dd = double of doublets, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant (J in Hz) and integration (number of protons). 13C NMR spectra were recorded on a 400 MHz Bruker AVANCE (100 MHz) or 600 MHz Bruker Ultrashield spectrometer (150 MHz). Data are reported as positions in parts per million (δ in ppm). Optical rotation data were acquired on a Perkin Elmer Model 341 Polarimeter using a 1 mL cell with a path length of 100 mm. Infrared (IR) spectra were recorded on a Perkin Elmer spectrum 100 instrument with a universal attenuated total reflection (ATR) attachment at room temperature. Wave numbers are reported in units of cm-1. Mass spectra were recorded using a Burker microTOF-Q II Electron Spray Ionization (ESI) Mass Spectrometer (MS).
Synthesis
C-undecyl Resorcin[4]arene Octa-ester, (2)13a14
To a stirring suspension of octol 1 (5.52 g, 5.0 mmol), oven-dried (110 °C) K2CO3 (7.6 g, 55 mmol) and a catalytic amount of sodium iodide (NaI) in dry acetonitrile (CH3CN) (50 mL) was added methyl-2-bromo acetate (3.9 mL, 40.5 mmol). The suspension was refluxed at 82 °C with stirring under a nitrogen atmosphere for 48 hours. After 24 hours another portion of methyl-2-bromoacetate (3.9 mL) was added. After cooling to room temperature, the mixture was filtered and the filtrate was extracted twice with diethyl ether (50 mL). The filtrate was concentrated under reduced pressure to give the crude product, which was re-crystallized from dichloromethane-methanol in 1:1 ratio. The precipitate was filtered and washed with methanol to yield the title compound as a white crystal. (6.5 g, 77 %); mp 90-93 °C [Literature mp 92-94 °C]; 1H NMR [CDCl3,400 MHz]: δ = 6.58 (s, 4 H, ArH), 6.20 (s, 4 H, ArH), 4.57 (t, 4 H, CH (methine)), 4.30 (s, 16 H, ArOCH2), 3.77 (s, 24 H, OCH3), 1.90 (q, 8 H, CH2(CH2)9CH3), 1.20-1.30 (m, 72 H, CH2(CH2)9CH3), 0.87 (t, 12 H, CH3) ppm; 13C NMR [CDCl3, 100 MHz]: δ= 169.81, 154.46, 128.51, 126.56, 100.76, 67.14, 51.91, 35.70, 34.52, 31.93, 30.01, 29.88, 29.79, 29.72, 29.39, 28.08, 22.69,14.10 ppm; FT-IR/ATR: 2920, 2851,1756,1729, 1610,1586,1501,1436,1405,1303,1210,1179,1105,1082,979,903, 850, 719, 584, 527 cm-1.
C-undecyl Resorcin[4]arene Octa-acid (3)14
To a stirring suspension of 2 (6.0 g, 3.57 mmol) in a mixture of 100 mL of ethanol and 50 mL of water was added potassium hydroxide (5.6 g, 99.96 mmol). The mixture was refluxed under a nitrogen atmosphere for 3 hours. The resulting mixture was concentrated under reduced pressure. The alkaline solution was acidified with 6 M HCl, and the suspension was extracted with ethyl acetate (100 mL x 3). The combined organic layers were dried over anhydrous Na2SO4. The solvent was removed in vacuo and the crude product was re-crystallized from methanol/ water in 1:1 ratio. After drying in vacuo the product was obtained as a white solid. (5.20 g, 93 %); mp 183-185 °C [Literature mp 180 °C]; 1H NMR [DMSO-d6,400 MHz]: δ =6.49 (s, 4 H, ArH), 6.35 (s, 4 H, ArH), 4.48 (t, 4 H, CH (methine)), 4.41 (d, 8 H, J= 16.3 Hz, OCH2-COOH), 4.23 (d, 8 H, J = 16.4 Hz, OCH2-COOH), 1.75 (q, 8 H, CH2(CH2)9CH3), 1.20-1.29 (m, 72 H, CH2(CH2)9CH3), 0.82(t, 12 H, CH3) ppm; 13C NMR [DMSO-d6, 100 MHz]: δ = 170.39, 154.00, 126.12, 125.29, 100.02, 65.89, 38.84, 34.93, 34.09, 31.29, 29.50, 29.25, 29.14, 29.06, 28.73, 27.68, 22.06, 13.85 ppm; FT-IR/ATR: 3195, 2920, 2850, 1719, 1612, 1587, 1499, 1435, 1407, 1298, 1184, 1127, 1105, 1072, 905, 812, 720, 665, 570 cm-1.
General Procedure for the Synthesis of Amino Acid Resorcin[4]arene Derivatives (4a-i)15
To a suspension of 3 (1.0 g, 0.64 mmol) in dry CH2Cl2 (30 mL) was added freshly distilled oxalyl chloride (1.1 mL, 12.80 mmol), and the mixture was refluxed for 18 hours under a nitrogen atmosphere. The unreacted oxalyl chloride and solvent were removed in vacuo, and the product obtained was dried under vacuum for 1 hour. Subsequently, it was dissolved in dry CH2Cl2 (20 mL), and slowly added to a cooled solution (0 °C) of amino acid ester hydrochloride (5.74 mmol) and triethyl amine (Et3 N) (1.35 mL, 9.80 mmol) in dry CH2Cl2(20 mL). The reaction mixture was allowed to warm up to room temperature and stirred under a nitrogen atmosphere for 18 hours. The solution mixture was treated with 1M HCl (30 mL), and the organic layer was separated, washed with water (30 mL), and brine (30 mL). The organic layer was dried over anhydrous Na2SO4. The solution was filtered, and the solvent was removed under pressure, yielding a sticky residue. The residue obtained was purified on silica gel chromatography using 3 % methanol in chloroform as a mobile phase. The fractions collected were concentrated with a rotary evaporator to give a white gum. These products were triturated with methanol to yield the title compounds as white solids.
Octa-glycine ethyl ester resorcinarene (4a)
Glycine ethyl ester hydrochloride (0.80 g, 5.74 mmol) was used as in the general procedure. The product was obtained as a white solid. (1.09 g, 76 %), Rf = 0.42 (3 % MeOH/CH3Cl), mp 97-100 °C; 1H NMR [DMSO-d6,600 MHz, 70 °C]: δ = 7.72 (t, J = 5.1 Hz 8 H, NH), 6.73 (s, 4 H, ArH), 6.57 (s, 4 H, ArH), 4.69 (t, J = 8.0Hz4H, CH (methine)),4.36 (d, J = 14.7Hz,8H,ArOCH2),4.27(d, J = 14.7Hz, 8 H, ArOCH2), 4.12 (q, J = 7.0 Hz 16 H, COOCH2CH3), 3.96 (dd, J = 5.1 Hz 8 H, Gly-CH2), 3.85 (dd, J = 5.8 Hz, 8 H, Gly-CH2), 1.81 (q, J = 6.5 Hz8H,CH2(CH2)9CH3), 1.20 (t, J = 8.5 Hz 24 H, COOCH2CH3), 1.22-1.27 (m, 72 H, CH2(CH2)9CH3), 0.84 (t, J= 7.5 Hz12H, CH3) ppm; 13C NMR [DMSO-d6,150 MHz, 70°C]: δ = 169.83, 168.82, 154.16, 127.08, 126.24, 100.84, 68.60, 60.89, 35.27, 35.14, 31.70, 29.90, 29.55, 29.51, 29.44, 29.10, 27.84, 22.44, 14.41, 14.19 ppm; FT-IR/ATR: 3414, 2920, 2851, 1757, 1731, 1664, 1586, 1501,1437,1406,1376, 1293,1191,1126,1085,1025, 904, 850, 720, 582 cm-1; MS (ESI-TOF) Calculated for C120H184O32N8Na [M+Na]+: m/z = 2273.2942, Found: m/z = 2273.2926.
Octa-alanine Methyl Ester Resorcinarene (4b)
L-Alanine methyl ester hydrochloride (0.80 g, 5.74 mmol) was used as in the general procedure. The product was obtained as a white solid. (1.05 g, 73 %); Rf = 0.45 (3 % MeOH/CH3Cl); mp 96-99 °C; [a]20D = -9.09 (c = 1.10, CHCl3); 1H NMR [DMSO-d6, 600 MHz, 70 °C]: δ = 7.82 (d, J= 7.3 Hz, 4 H, NH), 7.80 (d, J= 7.3 Hz, 4 H, NH), 6.73 (s, 4 H, ArH), 6.56 (s, 4 H, ArH), 4.67 (t, J = 5.0 Hz4H, CH (methine)),4.42 (q, J = 8.5 Hz 8 H, Ala-α H), 4.35 (q, J = 5.0 Hz 8 H, ArOCH2), 4.24 (q, J = 5.0 Hz 8 H, ArOCH2), 3.64 (d, J = 7.0 Hz 24 H, OCH3), 1.83 (q, J = 6.5 Hz8H,CH2(CH2)9CH3), 1.34 (t, J = 7.2 Hz 24 H, Ala-CH3), 1.20-1.28 (m, 72 H, CH2(CH2)9CH3), 0.84 (t, J = 6.0 Hz 12 H, CH3) ppm; 13C NMR [DMSO-d6, 150 MHz, 70 °C]: δ = 172.92, 172.84, 168.18, 168.09, 154.40, 127.19, 127.13, 126.27, 101.43, 68.80, 68.76, 52.27, 47.83, 47.77, 35.39, 35.13, 31.69, 29.88, 29.50, 29.41, 29.08, 28.09, 22.44, 17.46, 17.43, 14.18 ppm; FT-IR/ATR: 3408, 2923, 2853, 1741, 1676, 1613,1568,1521,1499,1436,1345,1293,1211,1157,1107,1055,987, 905, 851, 756, 720, 634, 541 cm-1; MS (ESI-TOF) Calculated for C120H184O32N8Na [M+Na]+: m/z = 2273.2942, Found: m/z = 2273.2969.
Octa-phenylalanine Methyl Ester Resorcinarene (4c)
L-Phenylalanine methyl ester hydrochloride (1.24 g, 5.74 mmol) was used as in the general procedure. The product was obtained as a white solid. (1.30 g, 71 %); Rf = 0.56 (3 % MeOH/CH3Cl); mp 58-61 °C; [α]20D = +24.24 (c = 1.10, CHCl3); 1H NMR [DMSO-d6, 600 MHz, 70 °C]: ddd = 7.77 (d, J = 8.2Hz, 4H, NH), 7.60 (d, J = 8.2 Hz, 4 H, NH), 7.04-7.15 (m, 40 H, Phe-ArH), 6.96 (s, 4 H, ArH), 6.33 (s, 4 H, ArH), 4.64 (q, J = 8.0 Hz 8 H, Phe-a h), 4.64 (t, J = 6.3 Hz4 H, CH (methine)), 4.28 (q, J = 7.1 Hz, 8 H, ArOCH2), 4.20 (q, J = 7.1 Hz, 8 H, ArOCH2), 3.59 (d,, J = 7.1 Hz, 24 H, OCH3), 3.11 (dd,, J = 6.0 Hz, 4 H, Phe-ArCH2), 3.04 (dd,, J = 6.0 Hz, 4 H, Phe-ArCH2), 3.03 (dd, J = 6.3 Hz, 4 H, Phe-ArCH2), 2.87 (dd, J = 6.3 Hz, 4 H, Phe-ArCH2), 2.17 (q, J =7.0 Hz, 8H, CH2(CH2)9CH3), 1.20-1.28 (m, 72 H, CH2(CH2)9CH3), 0.83 (t, J = 6.3 Hz, 12 H, CH3) ppm; 13C NMR [DMSO-d6, 150 MHz, 70 °C]: δ = 171.77, 171.73, 168.32, 168.27, 154.16, 137.27, 137.07, 129.36, 129.33, 128.67, 128.57, 128.52, 127.07, 126.87, 126.77, 126.35, 125.31, 100.43, 68.33, 53.59, 53.42, 52.23, 52.20, 37.39, 37.26, 36.07, 34.78, 34.59, 31.71, 30.95, 29.96, 29.59, 29.52, 29.43, 29.11, 27.86, 22.44, 21.42, 14.17 ppm; FT-IR/ATR: 3411, 3030, 2923, 2853, 1741, 1682, 1607, 1586, 1497, 1436, 1358, 1288, 1192, 1123, 1060, 905, 849, 815, 743, 699, 540, 490 cm-1; MS (ESI-TOF) Calculated for C168H216O32N8Na [M+Na]+: m/z = 2881.5446, Found: m/z = 2881.7059.
Octa-leucine Methyl Ester Resorcinarene (4d)
L-Leucine methyl ester hydrochloride (1.04 g, 5.74 mmol) was used as in the general procedure. The product was obtained as a white solid. (1.22 g, 72 %); Rf = 0.56 (3 % MeOH/CH3Cl); mp 61-64 °C; [a]20D = -20.00 (c = 1.05, CHCl3); 1H NMR [DMSO-d6, 600 MHz, 70 °C]: δ = 8.02 (d, J = 8.1 Hz, 4 H, NH), 7.79 (d, J = 8.1 Hz, 4 H, NH), 6.89 (s, 4 H, ArH), 6.53 (s, 4 H, ArH), 4.77 (t, J = 7.5 Hz, 4 H, CH (methine)), 4.47 (q, J = 3.56 Hz, 8 H, Leu-a h),4.46 (q, J = 7.1 Hz, 8 H, ArOCH2), 4.36 (q, J = 7.6 Hz, 8 H, ArOCH2), 3.62 (d, J = 17.5 Hz, 24 H, OCH3), 1.81 (q, J = 6.7 Hz,8H, CH2(CH2)9CH3), 1.45-1.67 (m, 24 H, Leu-CH and Leu-CH2), 1.20-1.28 (m, 72 H, CH2(CH2)9CH3), 0.87 (t, J = 7.5 Hz, 24 H, Leu-CH3), 0.84 (t, J = 7.1 Hz, 12 H, CH3), 0.77 (t, J = 7.2 Hz, 24 H, Leu-CH3) ppm; 13C NMR [DMSO-d6,150MHz, 70 °C]: δ = 172.83, 172.70, 168.36, 168.27, 154.32, 154.11, 127.38, 127.11, 126.41, 100.99, 68.81, 68.55, 52.18, 50.42, 35.82, 34.81, 31.68, 29.96, 29.55, 29.52, 29.50, 29.39, 29.08, 28.05, 24.83, 24.69, 22.98, 22.92, 22.42, 21.80, 21.72,14.15 ppm; FT-IR/ATR: 3415, 2924, 2853,1742,1679, 1613, 1585, 1523, 1498, 1437, 1368, 1275, 1196, 1154, 1104, 1056, 985, 902, 827, 721, 546, 466 cm-1; MS (ESI-TOF) Calculated for C144H232O32N8Na [M+Na]+: m/z = 2609.3198, Found: m/z = 2609.3148.
Octa-proline Methyl Ester Resorcinarene (4e)
L-Proline methyl ester hydrochloride (0.95 g, 1.75 mmol), was used as in the general procedure. The product was obtained as a white solid. (1.08 g, 69 %); Rf = 0.43 (3 % MeOH/CH3Cl); mp 65-68 °C; [a]20D = -80.00 (c = 1.00, CHCl3); 1H NMR [DMSO-d6, 600 MHz, 70 °C]: δ = 6.82 (br s, 4 H, ArH), 6.38 (br s, 4 H, ArH), 4.68 (t, J = 7.9 Hz, 4H, CH (methine)), 4.48 (br d, 16 H, ArOCH2), 4.38 (q, J = 5.66 Hz, 8 H, Pro-a H), 3.69 (d, J = 7.3 Hz, 24 H, OCH3), 3.56 (m, 16 H, Pro-δ H), 2.41(m, 8 H, Pro-/? H), 2.00-2.15 (m, 24 H, Pro-? H and g H), 1.90 (q, J = 4.3 Hz, 8 H, CH2(CH2)9CH3), 1.20-1.30 (m, 72 H, CH2(CH2)9CH3), 0.86 (t, J = 6.5 Hz,12 H, CH3) ppm; 13C NMR [DMSO-d6, 150 MHz, 70 °C]: δ = 172.61, 167.00, 154.85, 126.46, 99.99, 68.91, 68.82, 59.18, 51.95, 46.39, 41.08, 35.87, 34.89, 31.65, 29.80, 29.50, 29.49, 29.44, 29.37, 29.01, 28.85, 28.00, 24.94, 22.34, 14.01 ppm; FT-IR/ATR: 3473, 2923, 2852, 1740, 1645, 1499, 1433, 1343, 1294, 1171, 1126, 1043, 910, 842, 720, 540, 416 cm-1;MS (ESI-TOF) Calculated for C136H200O32N8Na [M+Na]+: m/z = 2481.4193, Found: m/z = 2481.4062.
Octa-tryptophan Methyl Ester Resorcinarene (4f)
L-Tryptophan methyl ester hydrochloride (1.46 g, 5.74 mmol), was used as in the general procedure. The product was obtained as a white solid. (1.29 g, 64 %), Rf = 0.42 (3 % MeOH/CH3Cl), mp 64-67 °C; [a]20D = +50.00 (c = 1.00, CHCl3); 1H NMR [DMSO-d6, 600 MHz, 70 °C]: δ = 10.42 (s, 4 H, NH-Indole), 10.32 (s, 4 H, NH-Indole), 7.63 (d, J = 7.56 Hz, 4 H, NH), 7.55 (d, J = 7.68 Hz, 4H, NH), 7.48 (t, J = 6.5 Hz, 8 H, H7-Indole), 7.29 (dd, J = 8.10 Hz, 8H, H4-Indole ), 7.02 (t, J = 7.4 Hz, 8 H, H5-Indole), 7.00 (t, J = 7.5 Hz, 8 H, H6-Indole), 6.90 (s, 8 H, H2-Indole), 6.80 (s, 4 H, ArH), 6.47 (s, 4 H, ArH), 4.71 (q, J = 7.1 Hz, 8 H, Trp-a H), 4.63 (t, J =8.0 Hz, 4 H, CH (methine)), 4.32 (q, J = 7.0 Hz, 8 H, ArOCH2), 4.26 (q, J = 7.2 Hz, 8 H, ArOCH2), 3.68 (d, J = 6.5 Hz, 24 H, OCH3), 3.11-3.25 (m, 16 H, Trp-β H), 1.83 (q, J = 7.7 Hz, 8 H, CH2(CH2)9CH3), 1.20-1.28 (m, 72 H, CH2(CH2)9CH3), 0.85 (t, J=6.5 Hz, 12 H, CH3) ppm; 13C NMR [DMSO-d6,150 MHz, 70 °C]:δ = 172.19, 172.14, 168.44, 168.35, 154.46, 139.53, 136.68, 136.65, 127.67, 127.60, 125.33, 123.97, 123.92, 121.36, 118.86, 118.39, 111.82, 111.79, 109.68, 109.56, 101.37, 68.85, 68.72, 53.41, 53.28, 52.12, 52.09, 35.39, 35.26, 34.80, 31.85, 30.96, 29.99, 29.65, 29.53, 29.44, 29.11, 28.02, 27.87, 27.84, 22.44, 21.42,14.19 ppm; FT-IR/ATR: 3403, 3056, 2923, 2852, 1738, 1671, 1585, 1496, 1435,1341,1287,1213,1179,1098,1059,928,860, 739,548,424 cm-1;MS (ESI-TOF) Calculated for C184H224O32N16Na [M+Na]+: m/z = 3194.6348, Found: m/z = 3194.6451.
Octa-serine (O-t-butyl) t-Butyl Ester Resorcinarene (4g)
O-t-Butyl-L-serine t-butyl hydrochloride (1.46 g, 5.74 mmol), was used in the general method. The product was obtained as a white solid. (1.19 g, 59 %), Rf = 0.62 (3 % MeOH/CH3Cl), mp 56-59 °C; [a]20D = +20.00 (c = 1.00, CHCl3); 1H NMR [DMSO-d6, 600 MHz, 70 °C]: δ = 7.27 (d, J = 8.2 Hz, 4 H, NH), 7.24 (d, J = 8.1 Hz, 4 H, NH), 6.73 (s, 4 H, ArH), 6.53 (s, 4 H, ArH), 4.63 (t, J = 8.0 Hz, 4 H, CH (methine)), 4.46 (q, J = 5.72 Hz, 8 H, Ser-a h), 4.36 (d, J = 14.70 Hz, 4 H, ArOCH2), 4.28 (d, J = 14.94 Hz, 4 H, ArOCH2), 4.25 (d, J = 14.88 Hz, 4 H, ArOCH2), 4.18 (d, J = 14.82 Hz, 4 H, ArOCH2), 3.70 (dd, J = 3.1Hz, 8 H, Ser-β CH2), 3.54 (dd, J = 4.6 Hz, 8 H, Ser-β CH2), 1.86 (q, J = 6.4 Hz, 8 H, CH2(CH2)9CH3), 1.40 (d, J = 3.5 Hz, 72 H, Ser-t-But), 1.20-1.28 (m, 72 H, CH2(CH2)9CH3), 1.09 (d, J = 3.8 Hz, 72 H, Ser-t-But), 0.86 (t, J = 6.5Hz,12H, CH3) ppm; 13C NMR [DMSO-d6,150 MHz, 70 °C]: δ 169.19, 168.22, 168.00, 154.69, 154.46, 154.39, 127.69, 127.58, 126.40, 101.57, 81.44, 81.35, 81.33, 73.12, 73.06, 73.03, 69.43, 69.04, 62.37, 62.28, 53.46, 53.40, 53.32,. 35.72, 35.25, 31.61, 30.70, 29.58, 29.49, 29.44, 29.32, 29.00, 27.89, 22.33, 22.31, 14.01 ppm; FT-IR/ATR: 3430, 2973, 2926, 2855, 1738, 1684, 1587, 1500, 1468, 1365,1293,1247,1192,1147,1098,1058,989,906,877,848,736,646, 566 cm-1; MS (ESI-TOF) Calculated for C176H296O40N8Na [M+Na]+: m/z = 3187.1331, Found: m/z = 3187.1433.
Octa-gulatmicacid (O -methoxy) Methyl Ester Resorcinarene (4h)
L-Glutamic acid dimethyl ester hydrochloride (1.22 g, 5.74mmol),wasusedasinthegeneralmethod.Theproductwas obtained as a white solid. (1.10 g, 60 %), Rf =0.46(3% MeOH/CH3Cl), mp 54-57 °C; [α]20D = -10.00 (c = 1.00, CHCl3); 1H NMR [DMSO-d6, 600 MHz, 70 °C]: δ = 7.66 (d d, J = 7.4 Hz, 8H, NH), 6.81 (s, 4 H, ArH), 6.55 (s, 4 H, ArH), 4.69 (t, J = 7.2 Hz, 4H, CH (methine)), 4.44 (q, J = 8.56 Hz, 8 H, Glu-α H), 4.32 (q, J = 8.56 Hz, 8 H, ArOCH2), 4.30 (q, J = 7.9 Hz, 8 H, ArOCH2), 3.70 ( s, 24 H, OCH3), 3.58 (d, J = 3.81 Hz, 24 H, OCH3), 2.36 (q, J = 4.2 Hz,16 H, Glu-g H), 2.13 (m, 8 H, Glu-β H), 1.96 (m, 8 H, Glu-β H), 1.86 (q, J=5.1Hz,8H,CH2(CH2)9CH3), 1.20-1.29 (m, 72 H, CH2(CH2)9CH3), 0.87 (t, J = 6.5 Hz, 12 H, CH3) ppm; 13C NMR [DMSO-d6, 150 MHz, 70 °C]: δ = 172.89, 172.87, 171.97, 171.93, 168.59, 154.47, 154.37, 127.26, 126.38, 101,26, 68.86, 68.65, 52.33, 52.32, 51.65, 51.62,51.45, 51.40, 35.11, 31.69, 30.12, 30.01, 29.95, 29.58, 29.51, 29.42, 29.09, 28.02, 26.74, 26.70, 22.44, 14.18 ppm; FT-IR/ATR: 3408, 2924, 2853, 1736, 1680, 1585, 1522, 1499, 1436, 1369,1293,1194,1170,1126,1056,985,901,824,721,638,556 cm-1; MS (ESI-TOF) Calculated for C144H216O48N8Na [M+Na]+: m/z = 2849.4632, Found: m/z = 2849.4642.
Octa-lysine (Νε-benzyloxyl) Benzyl Ester Resorcinarene (4i)
Ne-Cbz-L-lysine benzyl ester hydrochloride (2.34 g, 5.74 mmol), was used as in the general procedure. The product was obtained as a white solid. (1.88 g, 67 %), Rf =0.52(3% MeOH/CH3Cl), mp 57-60 °C; [a]20D = +7.69 (c = 1.26, CHCl3); 1H NMR [DMSO-d6, 600 MHz, 70 °C]: δ = 7.61 (dd, J= 7.8 Hz, 8H, NH), 7.26-7.36 (m, 80 H, Lys-ArH), 6.84 (s, 4 H, ArH), 6.61 (br t, 8H, Lys-e NH), 6.59 (s, 4 H, ArH), 5.13 (m, 16 H, Lys-cbz-CH2O), 4.77 (s, 16 H, Lys-Bn-CH2O,), 4.68 (t, J =7.2Hz,4H,CH (methine)), 4.44 (q, J = 6.1 Hz, 8 H, Lys-α H), 4.35 (q, J = 8.42 Hz, 8 H, ArOCH2),4.27 (q, J = 8.0 Hz, 8 H, ArOCH2), 3.10 (q, J = 5.2 Hz, 16 H, Lys-e CH2), 1.86 (q, J = 6.8 Hz, 8H, CH2(CH2)9CH3), 1.82 (m, 8H, Lys-S CH2), 1.78 (m, 8 H, Lys-S CH2), 1.39(m,16 H, Lys-δ CH2), 1.39 (m, 16 H, Lys-g CH2), 1.20-1.28 (m, 72 H, CH2(CH2)9CH3), 0.80 (t, J= 6.7 Hz, 12 H, CH3) ppm, 13C NMR [DMSO-d6, 150 MHz, 70 °C]: δ = 171.84, 171.82, 168.52, 168.44, 156.50, 154.76, 154.66, 137.87, 136.32, 136.29, 128.80, 128.42, 128.33, 128.13, 128.12, 127.96, 127.71, 126.49, 125.29, 101.92, 69.35, 69.14, 66.63, 66.60, 65.73, 52.43, 52.40, 49.05, 35.51, 34.75, 31.63, 31.55, 31.02, 29.99, 29.58, 29.50, 29.46, 29.39, 29.02, 28.05, 22.99, 22.33, 14.00 ppm; FT-IR/ATR: 3324, 3064, 3033, 2924, 2854, 1682, 1585, 1521, 1498, 1455, 1345, 1244, 1177, 1128, 1055, 1027, 910, 824, 735, 695, 576, 458 cm-1; MS (ESI-TOF) Calculated for C256H320O48N16Na [M+Na]+1/2: m/z = 2217.1575, Found: m/z = 2217.1494.
Supplementary material
The proton and carbon NMR data can be found in the online supplement.
Acknowledgements
We thank UKZN and NRF for funding of this project.
References
1 a) A.G.S. Hoegberg, Two stereoisomeric macrocyclic resorcinol-acetaldehyde condensation products, J. Org. Chem., 1980, 45, 4498^500; [ Links ] b) L.M. Tunstad, J.A. Tucker, E. Dalcanale, J. Weiser, J.A. Bryant, J. C. Sherman, R.C. Helgeson, C.B. Knobler and D. J. Cram, Host guest complexation. 48. Octol building-blocks for cavitands and carcerands, J. Org. Chem, 1989, 54, 1305-1312; [ Links ] c) E.U.T. Vanvelzen, J.F.J. Engbersen and D.N. Reinhoudt, Self-assembled monolayers of receptor adsorbates on gold - Preparation and characterization, J. Am. Chem. Soc., 1994, 116, 3597-3598. [ Links ]
2 a) C. Naumann, E. Roman, C. Peinador, T. Ren, B.O. Patrick, A.E. Kaifer and J.C. Sherman, Expanding cavitand chemistry: the preparation and characterization of n cavitands with n >= 4, Chem. Eur. J., 2001, 7, 1637-1645; [ Links ] b) K. Misztal, A. Sartori, R. Pinalli, C. Massera and E. Dalcanale, Design and synthesis of a cavitand pillar for MOFs, Supramol. Chem., 2014, 26, 151-156. [ Links ]
3 a) A. J. Wright, S.E. Matthews, W.B. Fischer and P.D. Beer, Novel resorcin 4 arenes as potassium-selective ion-channel and transporter mimics, Chem. Eur. J., 2001, 7, 3474-3481; [ Links ] b) E.K. Kazakova, A.V. Prosvirkin, V.V. Yanilkin, R. Froehlich and W.D. Habicher, A novel and effective strategy for the construction of "tube-like" double resorcin 4 arenes, J. Inclusion Phenom., 2003, 47, 149-153; [ Links ] c) P. Ogirala, S. Negin, C. Agena, C. Schaefer, T. Geisler, J. Mattay and G.W. Gokel, Properties of long alkyl-chained resorcin 4 arenes in bilayers and on the Langmuir trough, New J. Chem., 2013, 37, 105-111. [ Links ]
4 a) D.J. Cram, J.M. Cram, Container Molecules and Their Guests, Vol. 4, Royal Society of Chemistry, Cambridge, 1994; [ Links ] b) P. Timmerman, W Verboom and D.N. Reinhoudt, Resorcinarenes, Tetrahedron, 1996, 52, 2663-2704. [ Links ]
5 a) B. Botta, M. Cassani, I.D'Acquarica, D. Subissati, G. Zappia and G. Delle Monache, Resorcarenes: hollow building blocks for the host-guest chemistry, Curr. Org. Chem., 2005, 9, 1167-1202; [ Links ] b) W Iwanek, A. Wzorek, Introduction to the chirality of resorcinarenes, Mini-Rev. Org. Chem., 2009, 6, 398-411. [ Links ]
6 A. Casnati, F. Sansone and R. Ungaro, Peptido- and glycocalixarenes: playing with hydrogen bonds around hydrophobic cavities, Acc. Chem. Res., 2003, 36, 246-254. [ Links ]
7 a) I. Elidrisi, S. Negin, P.V. Bhatt, T. Govender, H.G. Kruger, G.W. Gokel and G.E.M. Maguire, Pore formation in phospholipid bilayers by amphiphilic cavitands, obc, 2011, 9, 4498-4506; [ Links ] b) I. Elidrisi, P.V Bhatt, T. Govender, H.G. Kruger and G.E.M. Maguire, Synthesis and NMR elucidation of novel amino acid cavitand derivatives, Tetrahedron, 2014, 70, 7057-7066. [ Links ]
8 a) W. Iwanek, M. Urbaniak, B. Gawdzik and V. Schurig, Synthesis of enantiomerically and diastereomerically pure oxazaborolo-benzoxazaborininone derivatives of resorcinarene from L-proline, Tetrahedron: Asymmetry, 2003, 14, 2787-2792; [ Links ] b) P. Shahgaldian, U. Pieles and M. Hegner, Enantioselective recognition of phenylalanine by a chiral amphiphilic macrocycle at the air-water interface: a copper-mediated mechanism, Langmuir, 2005, 21, 6503-6507; [ Links ] c) C.F. Dignam, J.J. Zopf, C.J. Richards and T.J. Wenzel, Water-soluble calix 4 resorcarenes as enantioselective NMR shift reagents for aromatic compounds, J. Org. Chem., 2005, 70, 8071-8078. [ Links ]
9 B. Botta, M. Botta, A. Filippi, A. Tafi, G. Delle Monache and M. Speranza, Enantioselective guest exchange in a chiral resorcin 4 arene cavity, J. Am. Chem. Soc., 2002, 124, 7658-7659. [ Links ]
10 a) L. Abis, E. Dalcanale, A. Duvosel and S. Spera, Structurally new macrocycles from the resorcinol aldehyde condensation - Configu-rational and conformational-analyses by means of dynamic NMR, NOE, and T1 experiments, J. Org. Chem., 1988, 53, 5475-5479; [ Links ] b) Y Aoyama, Y. Tanaka and S. Sugahara, Molecular recognition. 5. Molecular recognition of sugars via hydrogen-bonding interaction with a synthetic polyhydroxy macrocycle, J.Am. Chem. Soc., 1989,111, 5397-5404. [ Links ]
11 a) M.M. Conn and J. Rebek, Self-assembling capsules, Chem. Rev., 1997, 97, 1647-1668; [ Links ]b) D.M. Rudkevich and J. Rebek, Deepening cavitands, Eur. J. Org. Chem, 1999, 1991-2005; [ Links ] c) W. Sliwa and J. Peszke, Chemistry of cavitands, Mini-Rev. Org. Chem., 2007, 4, 125-142. [ Links ]
12 a) T. Fujimoto, C. Shimizu, O. Hayashida and Y. Aoyama, Solution-to-surface molecular-delivery system using a macrocyclic sugar cluster. Sugar-directed adsorption of guests in water on polar solid surfaces, J. Am. Chem. Soc., 1997, 119, 6676-6677; [ Links ] b) T. Fujimoto, C. Shimizu, O. Hayashida and Y. Aoyama, Ternary complexation involving protein. Molecular transport to saccharide-binding proteins using macrocyclic saccharide cluster as specific transporter, J. Am. Chem. Soc, 1998,120, 601-602; [ Links ] c) O. Hayashida, M. Kato, K. Akagi and Y. Aoyama, Interaction of sugar and anion in water via hydrogen bonding: Chain-length dependent agglutination of oligosaccharide clusters induced by multivalent anion binding, J. Am. Chem. Soc., 1999, 121, 11597-11598; [ Links ]d) O. Hayashida, J. Ito, S. Matsumoto and I. Hamachi, Preparation and unique circular dichroism phenomena of urea-functionalized self-folding resorcinarenes bearing chiral termini through asymmetric hydrogen-bonding belts, OBC, 2005, 3, 654-660. [ Links ]
13 a) J.R. Fransen and P.J. Dutton, Cation binding and conformation of octafunotionalized calix 4 resorcinarenes, Can. J. Chem., 1995, 73, 2217-2223; [ Links ] b) V.K. Jain, S.G. Pillai, R.A. Pandya, Y.K. Agrawal and P.S. Shrivastav, Molecular octopus: octa functionalized calix 4 resorci-narene-hydroxamic acid C4RAHAfor selective extraction, separation and preconcentration of U(VI), Talanta, 2005, 65, 466-475. [ Links ]
14 A. Bazzanella, H. Morbel, K. Bachmann, R. Milbradt, V. Bohmer and W. Vogt, Highly efficient separation of amines by electrokinetic chromatography using resorcarene-octacarboxylic acids as pseudo-stationary phases, J. Chromatogr. A, 1997, 792, 143-149. [ Links ]
15 a) F. Sansone, S. Barboso, A. Casnati, M. Fabbi, A. Pochini, F. Ugozzoli and R. Ungaro, Synthesis and structure of chiral cone calix 4 arenes functionalized at the upper rim with L-alanine units, Eur. J. Org. Chem, 1998,897-905; [ Links ] b) L. Frkanec, A. Visnjevac, B. Kojic-Prodic and M. Zinic, Calix 4 arene amino acid derivatives. Intra- and inter-molecular hydrogen-bonded organization in solution and the solid state, Chem. Eur. J., 2000, 6,442-453; [ Links ] c) M. Ree, J.S. Kim, J.J. Kim, B.H. Kim, J. Yoon and H. Kim, Cavitands bearing four fluorophores, Tetrahedron Lett, 2003, 44, 8211-8215. [ Links ]
16 a) A.G.S. Hogberg, Stereoselective synthesis and DNMR study of 2 1,8,15-22-Tetraphenyl 14 Metacyclophan-3,5,10,12,17,19,24,26-Octols, J. Am. Chem. Soc., 1980, 102, 6046-6050; [ Links ]b) S. Strandman, M. Luostarinen, K. Niemela, H. Tenhu and K. Rissanen, Resorcinarene-based ATRP initiators for star polymers, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4189-4201. [ Links ]
17 B.C. Gibb, A.R. Mezo, A.S. Causton, J.R. Fraser, F.C.S. Tsai and J.C. Sherman, Efficient coupling of amino-acid derivatives to rigid organic scaffolds - Model syntheses for de-novo proteins, Tetrahedron, 1995, 51, 8719-8732. [ Links ]
Received 25 September 2014
Revised 14 January 2015
Accepted 14 January 2014
* To whom correspondence should be addressed. E-mail: maguireg@ukzn.ac.za
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}