










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.68 Durban 2015
http://dx.doi.org/10.17159/0379-4350/2015/v68a2
RESEARCH ARTICLE
http://dx.doi.org/10.17159 /0379-4350/2015/v68a2
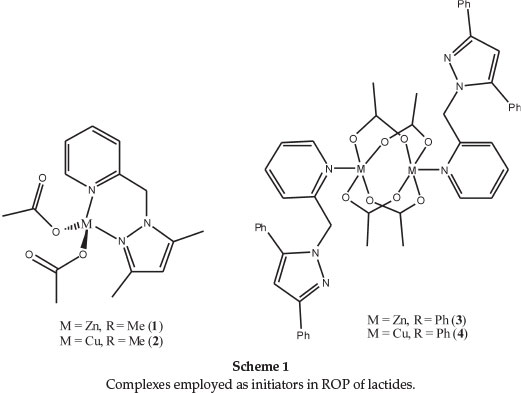
Ring-opening polymerization of lactides by (pyrazol-1-ylmethyl)pyridine Zn(II) and Cu(II) complexes: kinetics, mechanism and tacticity studies
Stephen O. Ojwach*; Thembisile P. Zaca
School of Chemistry and Physics, University of KwaZulu-Natal, Pietermaritzburg Campus, Private Bag X01 Scottsville, 3209 South Africa
ABSTRACT
The kinetics, mechanism and polymer microstructure studies of ring-opening polymerization (ROP) of lactides (LA) by Zn(II) and Cu(II) complexes of (pyrazolylmethyl)pyridine ligands, 2-(3,5-dimethylpyrazol-1-ylmethyl)pyridine (L1) and 2-(3,5-diphenylpyrazol-1-ylmethyl)pyridine (L2) is described. The complexes [Zn(Ac)2(L1)] (1), [Cu(Ac)2(L1)] (2), [Zn(Ac)2(L2)] (3) and [Cu2(Ac)4(L2)2](4) form active initiators in the ROP of D,L-LA and L-LA monomers. Generally Zn(II) complexes 1 and 3 exhibit higher activities compared to the corresponding Cu(II) complexes 2 and 4. Polymerization kinetics of D,L-LA show higher rates compared to the L-LA reactions. All the polymerization reactions follow pseudo first-order kinetics with respect to monomer while 1 shows second-order dependency on the polymerization reactions. Molecular weights of the polymers range from 813 g mol-1 to 9207 g mol-1 and exhibit relatively narrow molecular weight distributions between 1.2 to 1.6. While poly(D,L-LA) are predominantly atactic, poly(L-LA) are largely isotactic. All polymerization reactions proceed through coordination insertion mechanism followed by hydrolysis of the end groups.
Keywords: Zinc and copper complexes, lactide, polymerization, kinetics, mechanism.
1. Introduction
Biopolymers derived from renewable resources have attracted growing attention over the last decades as biocompatible and biodegradable alternatives to petrochemical-based plastics.1-5 An excellent example of such a biodegradable polymer is polylactide (PLA), which can be obtained by ring-opening polymerization (ROP) of lactide monomers. PLAs display desirable mechanical, optical and chemical properties such as optical transparency, ease of processing and ease of microbial decomposition or degradation. Due to these unique features, PLAs have wide range of applications in food packaging6a as well as in pharmaceutical industries, tissue engineering and drug delivery devices.6b,c
The ring-opening polymerization of lactides can be catalyzed by organometallic initiators,7 organocatalysts8 as well as enzymes.9 Organometallic-based catalysts have several advantages that include proper control of polymer stereo-regularity and molecular weight distribution.10-12 Furthermore, it is possible to carry out detailed kinetic and mechanistic studies of the polymerization reactions and gain fundamental knowledge on the effect of structure on catalyst activity and polymer micro-structure.13 Although tin(II) octanoate14 as well as several metal alkoxides15 are efficient catalysts in the ROP of lactides, design of well-defined catalysts that could produce PLAs with desirable molecular weight and polymer microstructure still remains a major challenge both in academia and in industry. One major drawback of most of these metal-based catalysts is their relative toxicity, which limits the application of the resultant PLAs in medical fields.6c,d In addition, the high cost of transition metal-based catalysts has dissuaded their industrial appeal.
To circumvent these two problems, numerous research groups are shifting towards inexpensive and less toxic metals such as calcium(II),16 magnesium(II)17 and zinc(II).18,19 We recently reported the use of pyrazolyl(methyl)pyridine Zn(II) and Cu(II) complexes in the ring-opening polymerization of ε-capro-lactone.20 In this current work, we explore the potential use of these breeds of complexes as initiators in the ROP of Meso-lactide (D,L-LA) and L-LA monomers. Detailed kinetics, mechanistic and polymer microstructure studies have been performed in order to elucidate the effect of catalyst structure and nature of monomer on the polymerization behaviour of these complexes.
2. Experimental
2.1. Materials
All air-sensitive manipulations were performed under argon using standard Schlenk line techniques. The Zn(II) and Cu(II) complexes were prepared according to our published proce-dures.20 Toluene and hexane solvents were distilled and dried from sodium/benzophenone mixture while dichloromethane was distilled from phosphorous pentaoxide. The monomers, D,L-LA and L-LA were purchased from Sigma-Aldrich and used as received. NMR spectra were recorded on a Bruker Ultra-shield 400 instrument at room temperature in CDCl3 (1Hat400 and 13C at 100 MHz). Mass spectrometry was recorded on a Waters Micromass LCT Premier Mass Spectrometer TOF with nano ACQUITY UPLC System. Gel permeation chromatography (GPC) analyses of the polymers were performed on a Waters 600E instrument.
2.2. Polymerization of Lactides
Ring-opening polymerization reactions were performed by introducing an appropriate amount of the complex, depending on the [LA]0/[I] ratio, in a Schlenk tube equipped with a magnetic stirrer under argon. LA (1.44 g, 0.01 mol) was then added and toluene (2 mL) was added to the Schlenk tube using a syringe and temperature was set at 110 °C. Kinetic experiments were carried out by withdrawing samples at regular intervals (approx. 0.1 mL) using a syringe and quickly quenched by rapid cooling into NMR tubes containing CDCl3 solvent. The quenched samples were analyzed by 1H NMR spectroscopy for determination of polymerization of LA to PLA. The percentage conversion of [PLA]t/([LA]t + [PLA]t) x 100, where [PLA]t and [LA]t are the concentrations of the polymer and LA monomer at time t, respectively, ([LA]t + [PLA]t, = [LA]0), was evaluated by integration of the peaks for LA (5.0 ppm, OCH signal) and PLA (5.2 ppm, OCH signal) according to the equation [PLA]/([LA]t + [PLA]t) = I52/(I52 + I50) where I50 is the intensity of the LA monomer signal at 5.0 ppm, and I52 is the intensity of the PLA signal at 5.2 ppm for the OCH protons.20 The observed rate constants, kobs, were extracted from the slopes of the lines of best-fit of the plots of ln[LA]0/[LA]tvs time. The polymers were purified by dissolving the crude products in CH2Cl2 followed by addition of cold methanol. A white precipitate was formed, which was isolated by filtration and dried to constant weight prior to analyses by NMR and GPC.
3. Gel Permeation Chromatography (GPC) Analyses
In a typical analysis, polymer samples were dissolved in 3,5-di-tert-butyl-4-hydroxytoluene (BHT) stabilized THF (2 mg/ml). The sample solutions were filtered via syringe through 0.45 μm nylon filters before analyses. The SEC instrument consists of a Waters 1515 isocratic HPLC pump, a Waters 717plus auto-sampler, Waters 600E system controller (run by Breeze Version 3.30 SPA) and a Waters in-line Degasser AF. A Waters 2414 differential refractometer was used at 30 °C in series with a Waters 2487 dual wavelength absorbance UV/Vis detector operating at variable wavelengths. Tetrahydrofuran (THF, HPLC grade, stabilized with 0.125 % BHT) was used as eluent at flow rates of 1 mL min-1. The column oven was kept at 30 °C and the injection volume was 100 μL. Two PLgel (Polymer Laboratories) 5 μm Mixed-C (300 x 7.5 mm) columns and a pre-column (PLgel 5 μm Guard, 50 x 7.5 mm) were used. Calibration was done using narrow polystyrene standards ranging from 580 to2x106 g mol-1. All molecular weights were reported as polystyrene equivalents and corrected by a factor of 0.58.
4. Results and Discussion
4.1. Ring-opening Polymerization of D,L-LA and L-LA Using Complexes 1-4 as Initiators.
Initial screening of complexes 1-4 (Scheme 1) as initiators in the ring-opening polymerization (ROP) reactions of lactides were performed at 110 °C in toluene using [M]/[I] ratio of 50. All the complexes 1-4 were found to display appreciable catalytic activities giving conversions above 90 % between 9-60 h (Fig. S1 in the online supplement). Having established the viability of 1-4 as catalysts in the ROP of D,L-LA and L-LA monomers, we performed detailed mechanistic and kinetics studies in order to understand the nature of the active species, influence of ligand motif and reaction conditions on catalyst activity and properties of the polymers obtained.
4.2. Kinetics of LA Polymerization Reactions
Kinetics of the polymerization reactions of lactide monomers catalyzed by complexes 1-4 were followed using 1H NMR spectroscopy. Table 1 gives a summary of the polymerization data. Plots of ln[LA]0/[LA]tvs. time for both monomers gave linear relationship consistent with a pseudo-first-order kinetics with respect to monomer for all the complexes (Fig. 1). The polymerization reactions thus follow simple pseudo-first-order kinetics with respect to lactide monomer as shown in Equation 1.

where k = kp[I]x, kp= rate of chain propagation and I = initiator; x = order of reaction
From Fig. 1, the rate constants for 1-4 were obtained as 0.328 h-1 (1), 0.066 h-1(2), 0.196 h-1(3) and 0.044 h-1(4) for D,L-LA and 0.157 h-1(1), 0.074 h-1(2), 0.107 h-1(3) and 0.039 h-1(4) for L-LA. The monomeric complex 1 was thus the most active while the dinuclear complex 4 was the least active. This trend contrasts our earlier reports where the binuclear complexes 3 and 4 were more active than their corresponding mononuclear analogues 1 and 2.20 Lower catalytic activities observed with increase in steric bulk of the pyrazolyl ligand in 1-4 could be largely apportioned to the steric crowding around the metal centre. Bridging acetate ligands in bimetallic complexes 3 and 4 could also play a role in blocking LA coordination to the active site hence lower activities. Mononuclear analogues 1 and 2 exhibit no such steric restrictions thus easier LA coordination to the metal centre. This observation is consistent with those of Carpentier and co-workers.21 Greater activities of Zn(II) initiators 1 and 3 in comparison to the Cu(II) catalysts 2 and 4 agree with literature reports.22
We also observed some influence of monomer stereo chemistry on the kinetics of the polymerization reactions. As depicted in Table 1, higher rate constants were reported for D,L-LA compared to L-LA reactions. For example, using catalyst 1, rate constants of 0.328 h-1 and 0.157 h-1 were obtained in the ROP of D, L-LA and L-LA monomers, respectively (Table 1, entries 1 and 5). Our findings are in tandem with recent reports by Buffet et at.23who reported 99 % and 66 % conversions for D,L-LA and L-LA monomers, respectively, under similar conditions. In general, rate constants observed for 1-4 were comparatively lower than most active initiators reported.24-26 For example, the mononuclear Zn-alkoxide complex reported by Williams et al.24 considered to be one of the most active zinc catalyst, exhibits a rate constant of 0.002 s-1 in the polymerization of L-LA. Despite the relative low activities of 1-4, they were found to be more active than the aluminium alkoxide catalyst reported by Zhong eta/.27whichdisplaysapparentrateconstantof0.902day-1inthe L-LA polymerization reactions.
4.3. Order of Reaction With Respect to Initiator
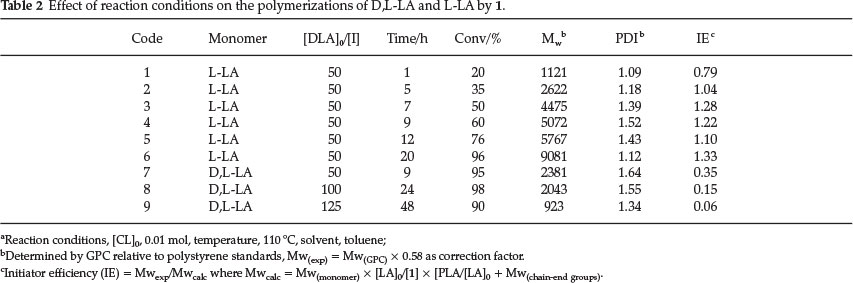
To assess the order of reaction with respect to initiator 1, the kinetics of the polymerization reactions were followed at different catalyst concentrations (Fig. S2) at constant D,L-LA monomer concentrations (Table 2). A plot of ln[kobs] versus ln [1] produced a linear relationship consistent with pseudo-first-order dependency of the reaction on [1] (Fig. 2). From the plot, the order of the reaction with respect to 1 was obtained as 2.32, indicative of a second-order dependency on 1. Fractional orders of the reaction with respect to initiator have been previously observed and are believed to arise from complicated aggregation of the active sites during polymerization reactions.28 Thus the overall rate law for D,L-LA polymerization by 1 can be represented as shown in equation 2. Attempts to determine the order of the reaction with respect to initiator 2 were unsuccessful due to longer induction periods which rendered the plots non-linear.

A linear plot of kobs vs [1] (Fig. 3) exhibited a non-zero intercept suggesting a threshold catalyst concentration of 0.02 mM, below which polymerization would not occur.29 William eta/.24 reported threshold values of 0.7 mM and 2.4 mM at 25 °C and 0 °C, respectively, for the Zn-alkoxide initiator. Our threshold concentration of 0.02 mM at 110 °C is thus significantly lower and could be apportioned to the relative thermal stability and tolerance to impurities of 1.
4.4. Molecular Weight and Molecular Weight Distribution of Polylactides (PLA)
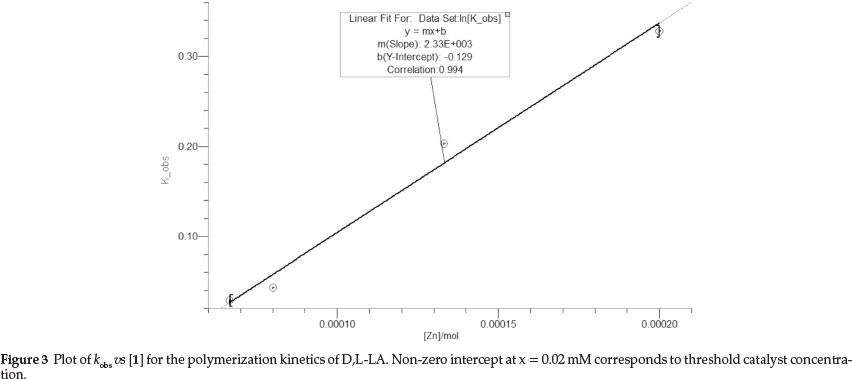
The experimental and theoretical molecular weights and molecular weight distributions of the PLAs were determined by GPC and 1H NMR spectroscopy, respectively (Tables 1 and 2). Lowtomoderateexperimentalmolecularweightsbetween813g mol-1 to 9207 g mol-1 were obtained. An interesting observation was the exceptionally high molecular weights of poly(L-LA) in comparison to poly(D,L-LA). For example, under similar conditions, molecular weights of 9081 g mol-1 and 2381 g mol-1 were obtained for L-LA and D,L-LA monomers, respectively (Table 1, entries 1 and 5). Indeed the experimental Mw of PLA obtained from L-LA were above the theoretical Mw, exhibiting initiator efficiencies (IE) of about 133 % compared to 35 % in D,L-LA polymerization reactions (Table 1 and Fig. 4). Reports of Buffet et al.30 sharply contrast our results as they recorded molecular weights of 18 000 g mol-1 and 10 750 g mol-1 for poly(D,L-LA) and poly(L-LA), respectively. Consistent with living polymerization behaviour, molecular weights increased with percentage conversion (Fig. 4). As an illustration, an increase in percentage conversion from 20 % to 96 % was marked by a concomitant increase in poly(L-LA) Mw from 1120 g mol-1 to 9081 g mol-1. The independence of molecular weight distribution on percentage conversion (Fig. 4) further augmented the living polymerization nature of 1.
We also studied the effect of monomer to catalyst ratio ([LA]/[1]) on polymer weight using catalyst 1. Contrary to our expectations and literature reports,31,32 we observed a decrease in molecular weight with increase in [LA]/[1] ratio (Table 2, entries 8-10). For instance, an increase in [LA]o/[1] from 50 to 125 resulted in decrease of molecular weight from 4105 g mol-1 to 1593 g mol-1, respectively. Low catalyst loading is expected to result in increased chain growth per active site. Thus at this stage, we are unable to unambiguously account for the observed trend. All the polymers obtained exhibited relatively narrow molecular weight distributions (1.09-1.64) compared to the wide distributions of 2.00-3.96 we reported in the polymerization of e-CL.20 The narrow PDIs coupled with the relatively high Mw observed in L-LA polymerization indicate absence/minimum transesterification reactions.33,34 However, low molecular weights of poly(D,L-LA) is consistent with the occurrence of intramolecular transesterification reactions.34 The narrow PDIs achieved for complexes 1-4 bearing acetate groups is in fact unusual as most acetate complexes give broad PDI in comparison to the alkoxide initiators.35
The nature of the metal centre also showed significant influence on the polymer molecular weight. Generally, zinc(II) complexes gave higher molecular weight PLAs than the analogous copper(II) complexes. For instance, poly(L-LA) molecular weights of 9081 g mol-1 and 2038 g mol-1 were obtained for complexes 1 and 2, respectively (Table 1, entries 5 and 6). Similar trends were observed in the polymerization of D,L-LA monomer. However, there was no significant steric influence on polymer molecular weight. Complexes 1 and 3 bearing methyl and phenyl groups on the pyrazolyl motif furnished molecular weights of 2381 g mol-1 and 2455 g mol-1, respectively (Table 1, entries 1 and 3).
4.5. Microstructural Analyses of Polylactides
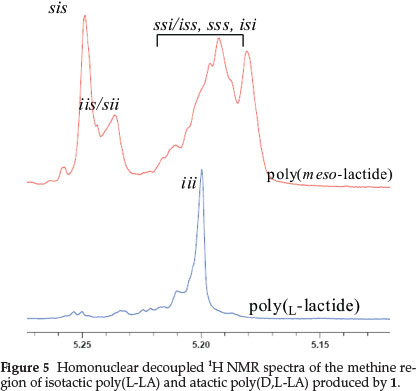
Poly(D,L-LA) and poly(L-LA) stereochemistry and tacticity were determined by homonuclear decoupled 1H{H} NMR and 13C NMR spectroscopy. Figure 5 represents the homonuclear decoupled 1H NMR and 13C NMR spectra of the methine regions of the polymers, respectively. From the 1H NMR spectra, it is evident that poly(L-LA) are predominantly isotactic while poly(-D,L-LA) are largely atactic (Scheme 2).36,3713C NMR spectra of both polymers were consistent with the interpretation of 1H NMR data.38 Production of atactic poly(D,L-LA) thus demonstrates lack of control of polymer stereoregularity by 1. This could be attributed to the absence of chirality in 1 as was demonstrated by optical N-heterocyclic carbene catalysts in the polymerization of D,L-LA to give predominantly heterotactic poly-mers.39 In addition, Ovitt and Coates have reported the use of chiral aluminium and yttrium alkoxide to produce heterotactic and syndiotactic PLAs from meso-lactide, respectively.40
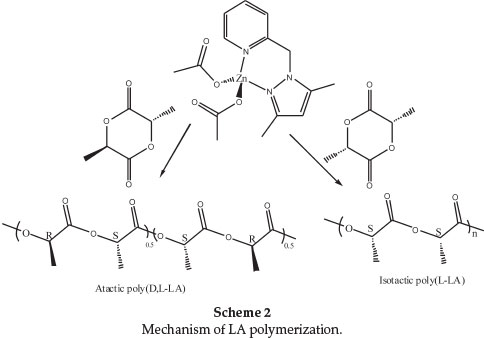
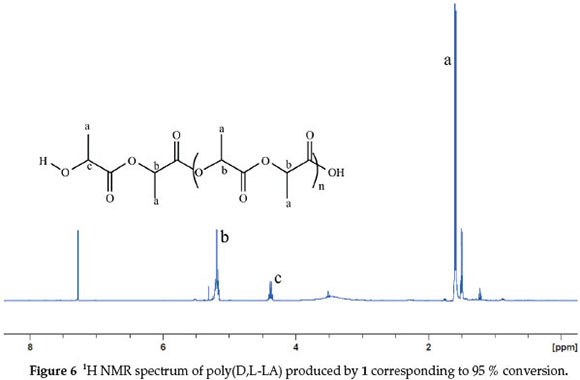
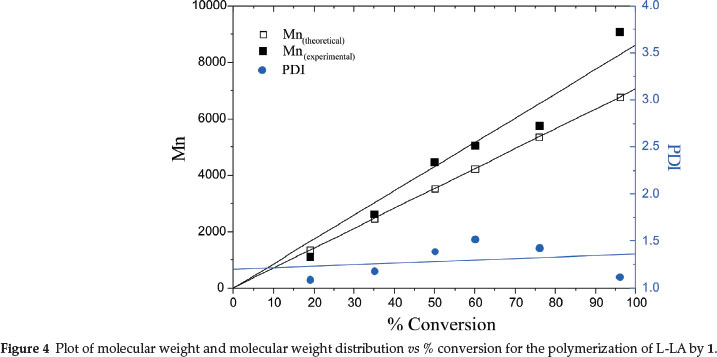
The ring-opening polymerization of LA by metal-based catalysts is likely to proceed either via coordination-insertion mechanism (CIM) or activated-monomer mechanism (AMM).7 In the CIM route, the polymer end chain bears the nucleophile (acetate in this case) on one end and the metal centre on the other end. However, chain transfer agents (water or alcohols) in the system might promote hydrolysis of the metal end to form an -OR end group.41 Polymerization through activated-monomer mechanism results in polymers bearing OR as end groups from an external nucleophiles (water or alcohols).42 To establish the mechanism of lactide polymerization by 1,1H NMR and ESI-MS spectra of the polymers obtained were analyzed. 1H NMR spectra of all the polymers revealed the absence of acetate methyl signals at about 2.00 ppm (Fig. 6). Similarly, no signals associated with ligand moiety in the complexes were observed. These observations are consistent with activated monomer mechanism. However, the linear dependency of polymerization kinetics with respect to [1] (Figs 2 and 3) strongly agree with coordination-insertion mechanism. Analyses of the ESI-MS spectra of poly(D,L-LA) exhibited signals corresponding to HO(C6H8O4)nHNa+ (Fig. 7). The presence of -OH end group could arise from adventitious water molecules. The -OH end groups could originate from adventitious water molecules that are capable of initiating hydrolysis of the polymer chain. These results are in agreement with those reported by Pilone et al.43 and Bourissou et al.34in which water molecules hydrolyzed the polymer end chains. From these data, the polymerization reactions by complexes 1-4 can thus be said to proceed through coordination insertion mechanism followed by hydrolysis of the acetate and complex end groups.
5. Conclusions
In summary, we have successfully shown that (pyrazol-1-ylmethyl)pyridine Zn(II) and Cu(II) complexes 1-4 formed active initiators for the ring-opening polymerization of D,L-LA and L-LA monomers. Both the nature of the metal atom and monomer influenced the catalytic behaviour of these compounds with Zn(II) complexes and L-LA displaying higher catalytic activities. The kinetics of the polymerization reactions were pseudo-first-order and second-order with respect to monomer and initiator 1, respectively. The polymerization reactions have been established to proceed through coordination insertion mechanism followed by hydrolysis of the acetate and complex end groups by water molecules. While poly(L-LA) were predominantly isoactactic, poly(D,L-LA) were mainly atactic indicating lack of control of polymer microstructure. Our current focus is on the design of catalyst systems that contain alkoxide initiating groups and stereogenic centres within the ligand framework to impart better control of polymer stereochemistry.
Supplementary Material
Supplementary Fig. S1 represents graphs of percentage conversions of D,L-LA and L-LA to PLA for catalysts 1-4 while Fig. S2 is a plot of ln[LA]0/LA]tvs time for 1 using different catalyst concentrations at constant monomer concentration of 0.01 mmol. GPC chromatograms for selected polylactides obtained are shown in Figs S3-S4. Figure S5 depicts the ESI-MS spectrum of crude poly(L-LA) obtained from 1.
Acknowledgements
The authors would like to thank University of KwaZulu-Natal for financial support.
References
1 A.P. Gupta and V Kumar, Eur. Polym J., 2007, 43, 4053-4073. [ Links ]
2 M. Vivas, and J. Contreras, Eur. Polym J., 2003, 39, 43-56. [ Links ]
3 F. Majoumo-Mbe, E. Smolensky, P. Lonnecke, D. Spasser, M.S. Eisen and E. Hey-Hawkins, J. Mol. Catal. A: Chem, 2005,240, 91-98. [ Links ]
4 L. Azor, C. Bailly, L. Brelot, M. Henry, P. Mobian and S. Dagorne, Inorg. Chem., 2012, 51, 10876-10883. [ Links ]
5 H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237-238. [ Links ]
6 C.S. Joseph, K.VH, Prashanth, N.K. Rastogi, A.R. Indiramma, S.Y. Reddy, K.S.M.S. Raghavarao, Food Bioprocess Technol., 2011, 4, 1179-1185. [ Links ](b)M. Minami, S. Kozaki, US patent 2003/6531615B2,2003. [ Links ] (c) B.D. Ulery,L.S. NairandC.T. Laurencin,J. Polym. Sci., PartB: Polym. Phys., 2011, 49, 832-864. [ Links ] (d) J.C. Middleton, A.J. Tipton, Biomaterials 2000,21, 8, [ Links ]
7 J. Wu, T.-L. Yu, C.-T. Chen and C-C. Lin, Coord. Chem. Rev., 2006, 250, 602-626.o [ Links ]
8 Y. Miao, Y. Phuphuak, C. Rousseau, T. Bousquet, A. Mortreux, S. Chirachanchai and P. Zinck, J. Polym. Sci. Part A: Polym. Chem., 2013, 51, 2279-2287. [ Links ]
9 S. Matsumura, Macromol. Biosci., 2002, 2, 105-126. [ Links ]
10 J. Cayuela, V Bounor-Legaré, P. Cassagnau and A. Michel, Macromolecules, 2006, 39, 1338-1346. [ Links ]
11 B.J. O'Keefe, M.A. Hillmyer and WB. Tolman, J. Chem. Soc. Dalton Trans., 2001, 2215-2224. [ Links ]
12 K.M. Stridsberg,M. Rynerand A.-C.Albertsson, Adv. Polym. Sci.,2002, 157, 41-65. [ Links ]
13 H. Du, X. Pang, H. Yu, X. Zhuang, X. Chen, D. Cui, X. Wang and X. Jing, Macromolecules, 2007,40, 1904-1913. [ Links ]
14 (a) A.C. Albertsson and I.K. Varma, Adv. Polymer Sci., 2002,157,1-40; [ Links ] (b) US Patent 5 258 488, 1993.
15 M.J. Stanford, P. Andrew and P. Dove, Chem. Soc. Rev., 2010, 39, 486-496. [ Links ]
16 M.H. Chisholm, J.C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717-6725. [ Links ]
17 B. J. Ireland, C.A. Wheaton and P.G. Hayes, OrganometaUics, 2010,29, 1079-1084. [ Links ]
18 C.-Y. Sung, C.-Y. Li, J.-K. Su, T.-Y. Chen, C.-H. Lin and B.-T. Ko, Dalton Trans., 2012, 41, 953-961. [ Links ]
19 V Poirier, T. Roisnel,J.-F. CarpentierandY. Sarazin, Dalton Trans., 2011, 40, 523-534. [ Links ]
20 S.O. Ojwach, T.T. Okemwa, N.W. Attandoh and B. Omondi, Dalton Trans., 2013, 42, 10735-10745. [ Links ]
21 M. Bouyahyi, N. Ajellal, E. Kirillov, C.M. Thomas and J.F. Carpentier, Chem. Eur. J., 2011,17, 1872-1883. [ Links ]
22 M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484-3504. [ Links ]
23 J-C. Buffet, J.P. Davin, T.P. Spaniol andJ. Okuda, New J. Chem, 2011,35, 2253-2257. [ Links ]
24 C. K. Williams, L.E. Breyfogle, S. Kyung Choi, W Nam, V. G. Young Jr., M.A. Hillmyer and W.B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350-11359. [ Links ]
25 H.-Y. Chen, B.-H. Huang and C.-C. Lin, Macromolecule, 2005, 38, 5400-5405. [ Links ]
26 H. Sun, J.S. Ritch and P.G. Hayes, Dalton Trans, 2012, 41, 3701-3713. [ Links ]
27 Z. Zhong, P.J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291-11298. [ Links ]
28 Y. Huang, W Wang, C.C. Lin, M.P. Blake, L. Clark, A.D. Schwarz and P Mountford, Dalton Trans, 2013, 42, 9313-9324. [ Links ]
29 C.M. Silvernail, L.Y. Yao, L.M.R. Hill, M.A. Hillmyer and W.B. Tolman, Inorg. Chem., 2007, 46, 6565-6574. [ Links ]
30 J.P. Davin, J.-C. Buffet, T.P. Spaniol and J. Okuda, Dalton Trans, 2012, 41, 12612-12618. [ Links ]
31 WM. Stevels, M.J. Ankone, P.J. Dijkstra and J. Feijen, Macromolecules, 1996,29, 6132-6138. [ Links ]
32 S. J. MClain, T.M. Ford and N.E. Drysdale, Polym. Prepr. (Am. Chem. Soc. Div. Polym. Chem.) 1992, 33, 463-464. [ Links ]
33 B.M. Chamberlain, B.A. Jazdzewski, M. Pink, M.A. Hillmyer and WB. Tolman, Macromokcuks, 2000, 33, 3970-3977. [ Links ]
34 D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993-9998. [ Links ]
35 Chamberlain, B.M., B.A. Jazdzewski, M. Cheng, D.R. Moore, T.M. Ovitt, E.B. Lobkovsky and G.W Coates, J. Am. Chem. Soc., 2001,123, 3229-3238. [ Links ]
36 K.A.M. Thakur, R.T. Kean, M.T. Zell, B.E. Padden and E.J. Munson, Chem. Commun, 1998, 1913-1914. [ Links ]
37 K.A.M. Thakur, R.T. Kean, E.S. Hall, J.J. Kolstad and E.J. Munson, Macromolecules, 1998, 31, 1487-1494. [ Links ]
38 W. Jiang, W. Huang, N. Cheng, Y. Qi, X. Zong, H. Li and Q. Zhang, Polymer, 2012, 53, 5476-5479. [ Links ]
39 A.P. Dove, H. Li, R.C. Pratt, B.G.G. Lohmeijer, D.A. Culkin, R.M. Waymouth and J.L. Hedrick, Chem. Commun., 2006, 27, 2881-2883. [ Links ]
40 T.M. Ovitt and G.W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686-4692. [ Links ]
41 G. Odian, Principles of Polymerization, 3rd edn., John Wiley & Sons, New York, 1991, pp. 550-551. [ Links ]
42 N. Ajellal, J.-F. Carpentier, C. Guillaume, S.M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363-8376. [ Links ]
43 A. Pilone, M. Lamberti, M. Mazzeo, S. Milione and C. Pellecchia, Dalton Trans., 2013,42, 13036-13047. [ Links ]
Received 31 March 2014
Revised 3 September 2014
Accepted 25 September 2014
* To whom correspondence should be addressed. E-mail: ojwach@ukzn.ac.za
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}