










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban ene. 2014
REVIEW ARTICLE
Peter Loyson
Chemistry Department, Nelson Mandela Metropolitan University, P.O. Box 77000, Port Elizabeth, 6031 South Africa
ABSTRACT
James Moir played a leading role in the South African Chemical Institute and was a pioneering organic chemist in South Africa during the early 1900s. He published widely on various organic topics but was especially noted for his work on the relationship between the colour of an organic substance and its chemical structure or constitution. After some initial research work in Scotland and London dealing with amarine and diacetonitrile, he arrived in South Africa in 1902. He first worked on derivatives of diphenol, oxidation products of benzidine, diphenylquinone and other compounds. His immense study of colour covered the years 1916-1929. He put forward theories on colour, which he applied to a whole range of phthaleins and related compounds, in order to see the effect of substitution on the spectrum of the compound. He introduced his numerical solution of colour, by which he could predict the band centre of absorption of many compounds. He applied this to a variety of different dyes and substituted products of phenolphthalein and fluorescein, relating colour to structure. He also studied the factors responsible for organic fluorescence, and doubly-linked diphenylene compounds. Finally he studied the structure of various flower pigments. This article explores his research output in the field of organic chemistry.
Keywords: Colour, phthaleins, history of chemistry, organic chemistry.
1. Introduction
James Moir (1874-1929) played an important role in the field of chemistry in South Africa in the early 1900s. His activities as a chemical analyst and his professional career have already been described in a previous paper.1 He was involved in many chemical investigations and was at the forefront of the development of chemistry, especially in the field of organic chemistry: in a series of 25 research papers published in the Transactions of the Royal Society of South Africa he reported the results of his investigations into the colour of organic compounds and the relationship between colour and chemical constitution or structure of these compounds; further work dealing with his organic chemistry research was published in the Journal of the Chemical Society, Transactions of the Chemical Society and in the pages of the Journal of the South African Chemical Institute. During his research he prepared a considerable number of organic compounds requiring complicated synthesis and he acquired a remarkable ability for this type of chemical work.2 This article traces his research contributions to organic chemistry.
Note: all of Moir's organic structures have been modernized, since he did not distinguish between an aromatic ring and a cyclohexane ring, but most of his naming has been retained in the article.
2. Early Work in Scotland and London
While studying at the University of Aberdeen he acted as a demonstrator in the Chemistry Department (1897-1900) and during this period he carried out research work on amarine under Prof. F. Japp. Amarine, 2,4,5-triphenylimidazoline, structure 1, is a poisonous crystalline substance obtained from the oil of bitter almonds.3 This research work dealt with the chemical composition and constitution of amarine, its supposed di-alkyl-and di-acyl-derivatives and of isoamarine.4 Amarine and its isomer, isoamarine, are important intermediates in the preparation of 1,2-diphenyl-diaminoethane.5

In 1900 he was awarded a scholarship and during this tenure at the Central Technical College in London he carried out investigations on some cyanohydroxypyridine derivatives of diaceto-nitrile.6 The latter compound could be formed through the action of Na on acetonitrile in the presence of a suitable diluent like ether. He showed that diacetonitrile was produced even in the absence of a solvent, as long as the Na was in excess. He also noticed, whilst crystallizing the diacetonitrile from hot water, that ammonia was evolved and that therefore a different substance crystallized from the liquid. The formation of this substance, sparingly soluble in all the ordinary solvents and beautifully crystalline, had already been observed before and its chemical formula was found to be C8H8ON2. The chemical structure of diacetonitrile had also been determined and it was suggested that the new substance could be formed from hydrolysis of the diacetonitrile to produce iso-cyanacetone. If two molecules of the latter compound lose one of water, a compound of the formula C8H8ON2 will be produced. Scheme 1 shows the changes.
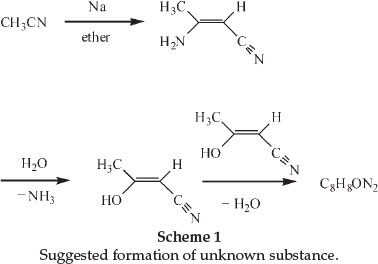
A German chemist named Holtzwart therefore suggested structure (A) for the compound C8H8ON2, it being the anhydride of cyanacetone. Another chemist, von Meyer, suggested an alternative structure (B).6
Moir's experiments led him to conclude that neither formula explained the extreme stability and inactivity of the substance, and after thorough investigations concluded that the substance was in reality 3-cyano- -lutidostyril or 2,4-dimethyl-3-cyano-6-hydroxy-pyridine (structure C). Dimethylpyridines were known as lutidines.7
-lutidostyril or 2,4-dimethyl-3-cyano-6-hydroxy-pyridine (structure C). Dimethylpyridines were known as lutidines.7

Using combustion analysis he confirmed the chemical formula of C8H8ON2 and characterized the compound, showing it to be phenolic in character and, being unusually stable, not to be affected by prolonged boiling with various reagents, even heating to 80 0C with fuming sulphuric acid. The first clue to the nature of the compound was obtained by heating it with zinc dust, yielding a distillate that smelled like pyridine. He hydrolyzed the compound by heating it with concentrated HBr in a sealed tube for 6 hours at 170 0C. This produced a large yield of a substance he called Ψ-lutidostyril, or 2,4-dimethyl-6-hydroxy-pyridine, by displacing CN in structure C by H, according to reaction 1 with the liberation of CO2 and NH3.
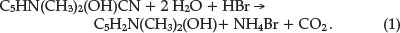
Brominating this latter compound gave 3,5-dibromo-Y-luti-dostyril (or 2,4-dimethyl-3,5-dibromo-6-hydroxy-pyridine), as expected from the parent structure. Nitrating the Ψ-lutidostyril gave 5-nitro-Y-lutidostyril, which upon reduction gave the corresponding amino compound.
In order to clarify the relative position of CN on the ring, he put forward two possible structures:
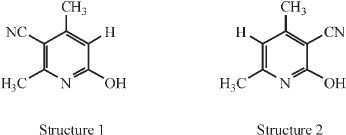
Both structures could be explained from a condensation of two molecules of the 'cyanacetone', depending on whether methyl or hydroxyl wandered in the process. Scheme 2 makes this clear.

The compound represented by structure 2 was already known then: it had been prepared by Guareschi in a way which left little doubt as to its chemical structure, namely by condensing acetylacetone, ammonia and cyanacetic ester (i.e. acetylaceto-namine and cyanacetamide) as shown in Scheme 3.8-10

He prepared the compound corresponding to structure 2 and compared it with the unknown compound C. The two compounds exhibited a remarkably close resemblance, both physically and chemically, and careful comparison was needed to determine that the two were in fact different: it was only in their derivatives that the two differed. The similarity in properties left no doubt that both were cyano--lutidostyrils and since the unknown compound is different from Guareschi's, it can only have the compound represented by structure 1, i.e. it is 3-cyano --lutidostyril.
Such a compound would only yield mono-derivatives; this was found to be the case, producing, upon bromination, the meta-bromo compound and, upon nitration, the meta-nitro compound. The best direct evidence of the position of the CN group in the unknown is afforded by the behaviour of the amino compound formed on reducing its nitro derivative. He found this to correspond to 5-amino--lutidostyril, as expected.
Finally, he attributed the unusual stability of this compound to 'the protective influence of the two methyl groups on every group which becomes imprisoned between them in the ring' and he gave other similar stable examples of benzene compounds such as the cyanoxylene, with the CN group in between two methyl groups and one with the CN group in between two bromine atoms on the ring. Modern interpretation would be the steric hindrance caused by the two methyl groups.11
During these investigations he also proved the non-existence of von Meyer's isomeric C8H8ON2 and converted Guareschi's isomeric compound (5-cyano-Y-lutidostyril) to the corresponding amide and carboxylic acid.
3. Work in South Africa
3.1. Initial Work
In 1902 James Moir arrived in South Africa and soon became involved in the mining industry : he investigated the chemistry of the extraction of gold from cyanide solutions and also became interested in the dust of mine air and its bearing on the causes of silicosis.2 In between his official duties as chemist with the Transvaal Department of Mines, he occupied his time with investigations on di-indigotin, diphenoquinone and its derivatives, and the quinonoid oxidation products of dianisidine and their polymerization and other topics.2,12
In 1907 he published his findings on new derivatives of diphenol (4,4'-dihydroxydiphenyl): he made the diphenol from benzidine by the di-azo reaction.13 He prepared and investigated the properties of diphenol dibenzoate and sulphonated the diphenol to give the 3,3'-disulphonic acid as the sole product.
A later investigation covers some remarkable oxidation products of benzidine,14 dealing with the dark-blue substances obtained by oxidizing benzidine with chromic acid, and with potassium ferricyanide; these substances resemble indigo in appearance, with very dark blue needles insoluble in most solvents except strong mineral acids and these reactions indeed were used as the best test for benzidine in nearly neutral solu-tions.14 They were described as simple chromate and ferricyanide of benzidine by their respective discoverers. Now he asserted that a colourless base must give a yellow chromate and ferri-cyanide, and that therefore these dark blue substances must be salts of an oxidation product of benzidine. He proposed that the oxidized base must be a hydrated form of diphenoquinonediimine, shown by Scheme 4, and the blue chromate salt would be diphenoquinonediimine chromate C12H12N2CrO4.

He showed similarly that the blue substance obtained using ferricyanide was actually a ferricyanide salt of diphenoquin-onediimine.
3.2. Colour and Chemical Constitution
In 1916 he started his immense study of the colours of organic compounds and over the next 13 years published 25 research articles dealing with various aspects of this topic in the Transactions of the Royal Society of South Africa. He also published a number of articles on this topic in other journals, where he proposed his theory of colour and criticized the theories of others.15-20
In his presidential address,21 upon becoming the President of the South African Chemical Institute for the second time in June 1925, he put forward his ideas about the chemical cause or origin of colour. His theory of colour depended on four postulates:
1. When light strikes any object, electrons are dislodged from such atoms as have free electrons; those of carbon are not, as a rule, free, but oxygen and nitrogen generally have free electrons, and after the dislodgement the atom which lost the electron is a positive centre of force.
2. These dislodged electrons may be caught by, or come under the attractive force, of a different positive centre.
3. If both the original and the positive centre occur in the molecule in such a way that their mutual distance does not alter much, the free electron belongs to both atoms and revolves around them both in an elliptic orbit which grazes the outsides of the two atoms. It is essential that the portion of the molecule concerned in colour-making be a practically rigid structure, without any internal rotation.
4. The rotation of the electron leads to the destruction of light of the particular wave-length that has the same number of vibrations per second as the number of rotations per second.
In addition to the postulates one has to mention that the elliptical orbit can only arise under rather limited conditions, such as the fact that the electron must be hit out at right angles roughly to the line joining the two positive centres; the result is that at a given moment only a few of the molecules are correctly orientated towards the light, and it requires a reasonable concentration of the coloured substance in solution to exhibit a colour.
The last step in the theory is that when a particular wavelength of white light is absorbed, the material exhibits the colour which is opposite or 'complementary' to the colour of the absorbed light. Thus if wave-length 400 nm is absorbed, which is the case when the elliptical orbit is small owing to the two active atoms being close together, the substance appears yellow (white light minus violet light); similarly if wave-length 650 nm is absorbed, when the atoms are 60 % further away than in the first case, the substance appears green (white light minus red light).
He concluded that colour was certainly of molecular and not atomic origin, and the height of the colour (i.e. the position of the absorption band) depended more or less directly on the distance between two active positive centres in the molecule, which could be two O atoms, or two N atoms, or one O and N atom, or one O (or N) and one unsaturated C atom.20,22
His research publications on colour of organic compounds can be broadly subdivided in the following: phthaleins and related compounds, numerical solutions of colour, dyes, substitution in phenolphthalein and fluorescein, organic fluorescence, doubly-linked diphenyl compounds and the colour of flowers. Each will now be described.
3.2.1. Phthaleins and Related Compounds
In his first study of the link between colour and structure of organic compounds he synthesized more than 50 phthaleins and other derivatives of the parent substance fuchsone (A) and analyzed their absorption spectrum in order to evaluate the effect of substituent groups and their positions within the structure.23 Phthaleins are chemical compounds formed by the reaction of phthalic anhydride with a phenol, from which certain synthetic dyes are derived. He started off with the simplest phthalein, which he called phenylphenolphthalein (B) which is colourless, but undergoes ring-opening, forming a purple-pink colour when its aqueous solution is made alkaline, giving a band centre of λ 560 nm, its structure given by (C). He asserted that the colour generated was due to the formation of the quinonoid structure within the ion.
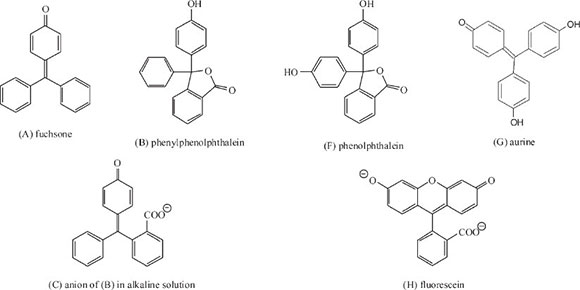
Then he moved on to examine benzaurine (D) or 4'-oxyfuchsone, which gives a reddish-purple colour in neutral solution (band centre at λ 570 nm); adding strong alkali moves the band centre to λ 585 nm, giving structure (E), which on warming becomes colourless, due to the collapse of the quinonoid structure. Benzaurine differs from the previous compound in having no o-COO- group, but has a p-OH group instead (Scheme 5).

He then investigated the effect of adding a second p-OH group using ordinary phenolphthalein (F) which is the p-OH derivative of (B) and aurine (G), the corresponding OH-derivative of benzaurine. In both cases the absorption band moves down the spectrum into the green: phenolphthalein gave λ 554 nm, whilst aurine gave λ 530. In the former case the effect of the extra -OH group was insignificant, but in the case of aurine, the difference was significant representing a near 10% rise in frequency: he attributed this to the almost perfect chemical symmetry present.
In his first paper he investigated 39 derivatives of phenolphthalein,23 listing the colour formed in alkaline solution, the centre of the absorption band, its frequency (1/1000 λ cm-1), the decrease in frequency and the cause for the decrease; in addition he also studied 13 derivatives of fluorescein (H). Derivatives included addition of one or more groups in particular positions within the structure of phenolphthalein or fluorescein. The following are some of the deductions he made:
a) Generally adding a substituent causes a displacement of the absorption band towards longer wavelengths.
b) The weight of the substituent is of little importance compared to its volume.
c) The shifting effect of the substituting groups on the absorption band is essentially due to the mutual interference of the two phenol rings: such groups as project much from the ring or are nearer to the central carbon cause a greater effect,
d) Groups on the phthalic ring have little effect.
e) Methoxyl groups have a remarkably large effect, suggesting its large volume.
f) The differences for the same substituent is not always similar, for example in the different Br-derivatives, which suggests that there is mutual action between the groups themselves tending to distort the quinonoid ring, which is the cause of the colour.
g) The effect of the O bridge in fluorescein (λ 499 nm) and its derivatives is profound and must be ascribed to the prevention of rotation of the phenol rings. He states that 'contrary to expectation, however, the frequency is raised (from 18.05 to 20.05 cm-1) in passing from phenolphthalein to fluorescein', whereas the usual effect of loading a molecule with a substituent is to lower the frequency as seen in the 39 derivatives of phenolphthalein.
Furthermore he discovered that the phthaleins and aurines, when dissolved in H2SO4, all give coloured solutions with definite absorption bands, giving an intensity of colour about five times as great as when dissolved in alkali. All phthaleins giving a blue colour in alkali give a pink colour in H2SO4. He analyzed the spectral data in concentrated H2SO4 for 13 phthaleins and 8 fluoresceins and derived a mathematical expression relating the frequency in alkaline solutions to the frequency in H2SO4 for the same phthalein.
In his next paper he examined the spectra of mixed phthaleins.24 He discovered that p-oxy-benzoyl-o-benzoic acid (see Scheme 6) combines with all sorts of phenols with the utmost ease (simply boiling the mixture) to yield dibasic mixed phthaleins containing one -C6H4OH group, as well as the residue of another phenol. In addition, the acid can combine with the most unexpected aromatic substances (phenol-esters, anhydrides and amines) to yield mixed phthaleins of a new type. He maintained that any ring compound with -OH or -NH2, either free or substituted, will react in this way, provided that it has a free para- or orthoposition. Scheme 6 below represents the combination of the simpler of the two cases, the para-position being the preferred one.

He summarized the spectral data for 18 dibasic mixed phthaleins, giving the band centre λ in alkali and in concentrated H2SO4. These phthaleins range from phenolthymolphthalein to phenol-a-naphthylamine-phthalein. Some of his observations were:
1) The acrylic group -CH:CH.COOH has practically the same effect on colour as the methyl group.
2) The pyridine ring has less effect on colour than the benzene ring.
3) When the phenolic -OH group has to take up the ortho-instead of the usual para- position to the central C, a greatly increased shift in λ follows.
The mixed amino-phthaleins are remarkably sensitive to excess alkali which easily bleaches their colour. The easy reaction of all these organic compounds with oxybenzoylbenzoic acid afforded a simple analytical test for phenols and amines, the position of the absorption band in the spectrum at once indicating which phenol (or amine) was used.
The following paper deals with derivatives of the unknown ortho-para-phenolphthalein.25 Ordinary phenolphthalein is a dioxy-derivative of diphenylphthalide, both -OH groups being para to the central C atom. He predicted that there should be a dioxydiphenylphthalide in which one of the -OH groups is ortho and the other para to the central C atom, and this, not being capable of forming an anhydride, should be soluble in alkali and should exhibit all the phthalein properties. He synthesized derivatives of this substance, including the substance itself, in which the ortho and para -OH groups are on different rings and which he therefore called o-p-phenolphthalein. As an example he prepared m-methyl-o-p-phenolphthalein (phenolparacresol-phthalein) and examined its spectrum. He prepared this from paracresol and p-oxybenzoylbenzoic acid using ZnCl2 as shown in Scheme 7.
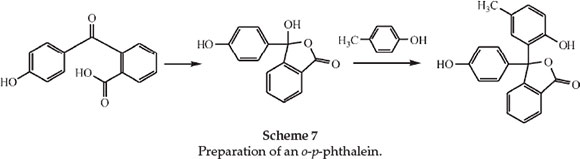
The phenolparacresolphthalein dissolved in alkali with an indigo colour, giving a band centre at λ 572 nm just the same as that for the ordinary orthocresolphthalein. Dissolved in concentrated H2SO4 it gave an intense salmon-orange shade at λ 487 nm. Further derivatives of o-p-phenolphthalein were made for spectroscopic observation.
Then he discovered that p-oxybenzoylbenzoic acid yields a phthalein-like substance when heated alone to over 200 0C or at a lower temperature when heated with a little H2SO4. He admitted that someone else had recently published the same observation but they incorrectly stated that the substance obtained was phenolphthalein.26 The new phthalein was pink-violet in alkali at λ 560 nm, whilst in H2SO4 it had λ 501 nm. Its spectrum was very similar to that of phenol-o-cresolphenolphthalein.
He also discovered a new property of phthaleins: it is possible to find a particular strength of alkali in which any phthalein gave a colourless solution in the cold, but which became coloured on heating to near the boiling point and again fades on cooling and allowing to stand. For ordinary phenolphthalein the alkali concentration was slightly over 1 N in strength. Tetraiodo-phenol-phthalein required much weaker alkali to exhibit this phenomenon, and a-naphtholphthalein required about 2 N alkali. He was puzzled about this behaviour.
In a further paper dealing with direct substitution products of ordinary phenolphthalein,27 he showed how he could predict the band centre of a particular phthalein. For example a-naphthol-thymolphthalein, blue-green in alkali at λ 633 nm offers an interesting case: he showed that the effects of the substitution are additive, i.e. the colour and position of the adsorption band of very complex phthaleins could be predicted from data of the simpler ones. a-Naphthol-thymolphthalein [structure (c)] can be looked on as either thymol-phenolphthalein [structure (a)] to which the butylidene group has been added (converting phenol into naphthol), or as a-naphtholphenol-phthalein [structure (b)] to which the methyl and isopropyl groups have been added (converting phenol - in the other ring -into thymol).

Now in the first case, the value of the butylidene group can be ascertained by comparing the spectrum of a-naphthol-phenol-phthalein with that of phenolphthalein. The central wavelengths of these substances are 607 nm and 554 nm, so that the single butylidene group which makes the difference between them, has raised the wavelength of the band by 9.5 %. Applying therefore a correction of +9.5 % to the figure for thymol-phenolphthalein (λ 578 nm) one gets 1.095 x 578 = λ 633 nm as the calculated value for a-naphthol-thymolphthalein. In the second case in the same way, he took the experimentally determined value for a-naphthol-phenolpthalein (λ 607 nm) and multiplied it by the ratio indicating the conversion of one phenol group into a thymol group (viz. 1.044 or 578/554): this gave a second theoretical value for the band centre in the supposed unknown a-naphthol-thymolphthalein, viz. 607 x 1.044 = λ 634 nm. The observed value is λ 633 nm, showing that both predictions were accurate.
Part 6 of his series dealt with UV spectra of the phthaleins.28 From previous data he suspected the phthalein bands seen in H2SO4 solution to be due to the 'loading' with H2SO4 of bands in the UV, which thus were brought into the visible spectrum. Also by using a strong solution of alkaline phenolphthalein its suspected UV band could be so broadened that a portion of it became visible between λ 360 and λ 400 nm.28
His observations were confirmed in 1917 and extended by photographic means by workers from Cornell University in America,29 but two new UV bands in alkaline phenolphthalein were described, lying at λ 370 nm and 277 nm, and in addition, their measurement of the visible band was the same as his (λ 554 nm). He noted that these wavelengths were in the ratio 1/2, 1/3, 1/4 and consequently the real fundamental absorption of alkaline phenolphthalein lay in the IR at λ1109 nm (frequency 9.02 x 103 cm-1); also the visible band was the first harmonic (λ 554 nm, frequency 18.05 x 103 cm-1), and the barely visible band in the extreme violet was its second harmonic (λ 370 nm, frequency 27.07 x 103 cm-1) whilst the band at λ 277 nm, frequency 36.1 x 103 cm-1, was its third harmonic.
In the case of higher phthaleins (e.g. thymolphthalein), the same frequency ratio of 2:3:4 was observed, the wavelengths being λ 626 nm, λ 417 nm and λ 313 nm. He showed that the wavelength of the violet absorption band was 2/3 that of the visible one.
He introduced a simplified formula y = x - 7, where x was the visible frequency of the particular phthalein in alkali and y that in H2SO4. Thus for phenolphthalein, x was 27.03 (1000/370) cm-1, and therefore y was 20.03 cm-1 (observed 1000/499 = 20.04 cm-1).
For thymolphthalein, x = 25.13 cm-1 (from 10000 divided by 2/3 of 597), hence y is calculated at 18.13 cm-1 (observed 18.18 cm-1). The constant 7 in the formula represented the 'loading' of the molecule with the constant molecular mass of H2SO4, or more probably the constant atomic volume of that substance as previously suggested. He showed that the data were not related to the molecular mass, and suggested that the molecular volume was responsible for cause of the position of the absorption band in each case.
A further paper in the series covered an empirical law of colour change applied to halogenated derivatives of phenolphthalein and fluorescein.30 Using halogen derivatives of the two parent substances he showed that the laws connecting the change in colour with the number and nature of the halogen substituents were quite simple and regular, so that all experimental results could be calculated and the spectrum of the unknown substances predicted from Equation (2):

where n = frequency (10000/1) of the band of the halogenated derivative, n0 = the frequency of the parent substance (18.05 cm-1 for phenolphthalein, 20.27 cm-1 for fluorescein), m the number of halogen substituents and N the atomic number of the halogen (17 for Cl, 35 Br and 53 for I). His data gave results in close agreement with the formula.
In the formula the factor involving the atomic number is small; when one neglects it, the additive law given in previous work was obtained, addition of wavelength being the same as subtraction of frequency. The formula held for fluorescein derivatives as well as phthaleins.
He applied Equation (2) to di-halogen derivatives, with two possible isomers, which he synthesized and examined. He even predicted the absorption band of the only trichloro-phenol-phthalein possible to be at λ 576 nm.
He then examined what the atomic number must be for compounds that are not halogenated: he suggested the sum of the atomic numbers of the constituents, i.e. methyl CH3 = 9, isopropyl = 25. With this formula he obtained excellent results in predicting the λ value for o-monomethylphenolphthalein (561 vs. 562 nm).
In view of these results, however, he had to abandon his previous suggestion that the molecular volume was the main factor in colour change, since such different groups as methyl, isopropyl, Cl, Br and I produced about the same effect.
3.2.2. Numerical Solutions of Colour
Part 10 of the series dealt with a general numerical solution of the colour constitution problem.31 He had shown before that the colour phenomenon is additive, i.e. each group present acts in colour change almost independently of other groups and of the rest of the molecule. He now found that each combination of group and position, e.g. o-methyl, p-hydroxyl, etc., can be assigned a colour-factor by means of which the effect on colour of the substitution of the group for H in the given position can be calculated. Even if five or six groups are present it is found that by multiplying the wavelength of the parent substance by all the factors of the groups present in the coloured substance a result is obtained which tallies closely with observation.
The parent substance was an imaginary compound, p-phenyl-enediphenylmethane (structure 2) or the anhydride of triphenylcarbinol. He called it fuchsene, it being the hydrocarbon of fuchsone.
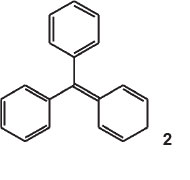
He calculated its fundamental 1 by multiplying that of benzaurine (553 nm, its dioxy-derivative) by the square of the ratio of the wavelengths of benzaurine and aurine (534 nm, its tri-oxyderivative), i.e.
X = 553 X 5532/5342 = 593 nm.
He now introduced various colour factors, the first one being the p-OH factor, taking the ratio of the wavelengths for aurine/benzaurine, its value being 534/553 = 0.9657. The theoretical λ for fuchsone (oxyfuchsene), if it were to dissolve in alkali, would thus be 593 X 0.9657 = λ 572.5 nm, that for oxyfuchsone or benzaurine (oxyoxofuchsene) is 593 X (0.9657)2 or 1 553 nm, and that of aurine ( dioxyfuchsone) is 593 X (0.9657)3 = λ 534 nm.
The next factor is the p-amino factor, the value being 0.972. He goes on to deal with diamino derivatives, which agree well. The two ratios are next combined: the theoretical λ for oxyfuchsoni-mine or aminofuchsone is 593 X 0.9657 X 0.972 = λ 556.5 nm (observed: 558 nm), that for diamino fuchsone is 593 X 0.9657 X (0.972)2 = λ 541 nm (observed: 541), and that for dioxyfuchsoni-mine is 593 X (0.9657)2 X 0.972 = λ 537.5 nm. He made this new substance and observed λ 538 nm.
The third factor is the N-methyl factor which is 1.0245. From this the colours of all the violet and green dyes of the series could be calculated. He now combined the second and third factors giving fourthly an N-methylamino factor, which is 0.9965, and fifthly a N-dimethylamino factor which is 1.021. Applying these, the theoretical colour of fuchsonedimethylimine is 593 X 1.021 = 605.5 nm. Numerous other examples are given.
In his next study he applied the formula 1 = 554 + 7.091 m + 0.091 m2 to calculate the wavelength of the ortho-substituted bromophenolphthalein,32 where m is the number of ortho-bromine atoms present in the brominated phenolphthalein, and where 554 is the wavelength for the unsubstituted phenolphthalein. If m is less than 5, the wavelength may be taken to be 7.5 units higher than 554 for each ortho-bromine atom present. Thus 561.5, 569, 576.5 and 584 are the wavelengths of (ortho) mono-, di-, tri- and tetra-bromophenolphthalein.
This treatment was limited to ortho-bromination on the phenol rings of phenolphthalein. Expanding this treatment to include substitution in any position on any of the three rings of the alkaline phenolphthalein structure, he then proceeded to prepare phthaleins derived from the reaction of appropriately brominated phthalic acids with substituted phenols and he showed how the position of the bromine atom(s) on the rings affected the colour of the compound. This enabled him to predict the colour of any polybromo-derivative. Numerical values are assigned to various locations on any of the three rings of the structure of phenolphthalein, shown in structure 3, and these values are then simply added to 554 to obtain the wavelength of absorption of the brominated phenolphthalein.
This treatment is applied to mono-, di-bromo-, tri-bromo-, tetra-bromo-, penta-bromo-, hexa-bromo-and finally hepta-bromophenolphthalein. Agreement between calculated and observed wavelengths was excellent.

In Part 12 of the series on Colour and Constitution, he calculated the colour from the tautomeric theory.33According to a new theory put forward by E.R.Watson,34 the depth of colour, or the displacement towards longer wavelengths, depended on the length of continuous tautomeric change or alteration of single and double bonds in the molecule. He applied this theory to his observations of wavelength (absorption centres) of most of the common dyes, to see if numerical values in wavelength can be assigned to each possible kind of tautomerism and each appending group, which could then be added up to give a result approximating that observed for the dye. This he found to be the case. He gave numerous examples where the calculated wavelength is very close if not equal to the observed one.
3.2.3. Dyes
He then considered the case of the three symmetrical aniline dyes, aurine, parafuchsine and crystal violet, which have always been a puzzle to the colour chemist, since their colours are unexpected. He used his theory and allocated numerical values to confirm the colour of each, for example for aurine in water he obtained 542 nm (observed: 543 nm); with crystal violet he predicted a value of 598 nm (observed: 596 nm). He also predicted what indigo should be, since it does not dissolve in water.
In Part 13 of the series of publications, he calculated the colour of the monocyclic dyes.35 A typical monocyclic dye would be dimethylaminobenzhydrol or PhCH(OH)C6H4NMe2. A typical dicyclic dye would be Ks-dimethylaminobenzhydrol, structure 4, then commonly called 'Michler's hydrol'.
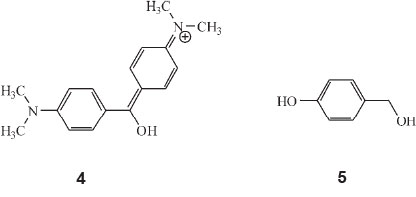
He now discovered that the colours of the monocyclic dyes (which are all low, viz. below λ 500 nm) can be calculated by a factorial scheme similar to that used previously. Starting off with the parent substance p-oxybenzylalcohol (oxyphenylcarbinol), structure 5, he allocated various factors to calculate colour: A, the phenyl factor for replacing H in carbinol by Ph, 1.135; B, the o-carboxyl factor for converting a phenylcarbinol into a phthalein, 1.060; C, the phthalein factor, orAxBfor replacing H by C6H4CO2H, 1.203, and others.
Armed with these data he calculated the colours of the known monocyclic coloured substances and predicted the colours of the unknown members of the series. Examples are:
Fuchsone: this is the diphenyl derivative of the parent substance. Its calculated λ was therefore 290 A2 = 290 x 1.1352 = 374 nm. A value of 380 nm had been reported in alcohol, but he maintained values for water were always six units less.
para-Oxybenzhydrol is the monophenyl derivative and gives λ = 329 nm. It had up to then not been observed.
Mono-oxydiphenylphthalide (phenylphenophthalein) is the ortho-carboxylic acid of fuchsone. Its calculated colour is 374 (for fuchsone) multiplied by 1.060 = λ 397 nm. He observed 395 nm in water.
His next paper covered dicyclic dyes.36 He worked out all the factors whereby the colour of any dye made up of two ionizable rings with either one or two linkages,can be calculated. Dioxybenzhydrol, structure 6, HOC6H4CH(OH)C6H4OH is an example of a singly-linked dicyclic dye, whilst dioxy-xanth-hydrol, structure 7, is an example of a doubly-linked dicyclic dye. Mono-oxybenzhydrol on the other hand is 'monocyclic' having only one ionizable ring.
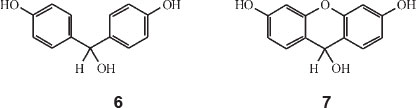
He started off with the assumption that the wavelength of an electron orbit going around two phenol rings is about λ 1380 nm and that this orbit was contracted by linkage of the rings always in the same proportion for the same linkage, whatever other groups are present. Each linkage thus had a colour factor (less than unity) which when multiplied by 1380 gives the colour of the corresponding dioxyphenyl dye.
A table of linkage factors, including oxidized linkages is shown below:
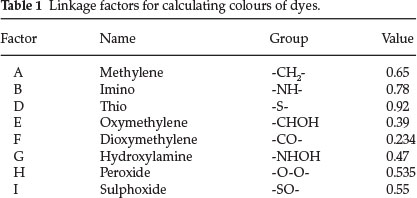
All of the 27 ring colour factors published in Part 10 could be used to extend the theory from the phenolic dyes to other classes of dicyclic dyes. He gave 15 examples of singly linked and a whole list of doubly-linked dicyclic colours.
Singly linked - One example is dioxybenzhydrol: calculated value for λ is 1380 x factor E = 1380 x 0.39 = 538 nm (observed: 539 nm). For phenolphthalein: same as above with a benzoic acid group attached; from previous work in Part 10, the factor for the latter is phenyl multiplied by o-carboxyl = 1.026 x 1.002 = 1.028. Calculated λ for phenolphthalein is 1380 x 0.39 x 1.028 = 553 nm (observed: 554 nm).
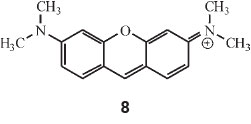
Regarding doubly-linked dicyclic colours he started off with pyronine, which he calculated by multiplying λ 1548 (for the two dimethylaniline rings) by factors C and E, i.e. 1548 x 0.89 x 0.39 = 536 nm (observed: 542 nm in alcohol).
Dioxyxanthhydrol is pyronine (structure 8) with 2 OH groups instead of 2 NMe2 groups: hence 1380 x 0.89 x 0.39 = 478 (observed: 479 nm). This is the parent substance of fluorescein.
Part 17 examined the azo dyes and other monocyclic colours.37 He used a diffraction grating specially mounted on a thin cover-glass, such as used in microscopy, to photograph absorption bands even down to λ = 320 nm and thus also study colourless substances. Alkaline p-oxyazobenzene in water immediately showed not one broad band at λ = 420 nm, as previously believed, but two overlapping bands at λ 433 nm and λ 395 nm. Further investigations showed that all the members of the family show this phenomenon. They are all indicators, which change colour if made acid or alkaline, thus giving three kinds of spectra according to pH conditions. Each substance has therefore six bands and he now proceeded to calculate each of these.

For example p-oxyazobenzene (structure 9) has λ 433 nm and 395 nm in alkaline, λ 490 nm and 463 nm in acid and λ 340 nm and 315 nm when neutral. Each of these represents a different chemical constitution. To calculate these bands, he started off with the observed λ of 287 nm for the phenate ion C6H5O- in water and allocated colour factors for certain additional structural components and any oxidation. For example the a-methylene (-CH2-) interposition colour factor is 0.92, the a-imino (-NH-) 1.09, the b-imino 1.22, the a-oxidation factor 1.10, and the b-phenyl substitution factor 1.03. Using these factors and others, one could calculate the bands of azobenzenes and many other related compounds.
The method is to multiply together all the factors taken from the formula of the substance, the result being the wavelength of the band centre corresponding to the particular structure. For example for the p-oxyazobenzene structure, PhNHNOHC6H4O-, 287 (phenate) X 1.09 (a-imino) X 1.10 (a-oxidation) X 1.22 (b-imino) X 1.03 (b-phenyl) gives 432 nm. He gives tables of oxy-compounds, monocyclic amino compounds and dimethylamino-compounds, all giving good agreement for their acid species.
A later paper (Part 24) examined the triphenylcarbinol or 'aniline' dyes.38 He synthesized the complete series of possible diphenylcarbinol and triphenylcarbinol dyes, all 23 of them! These 23 were found to possess no less than 75 absorption bands amongst them; he tackled the 'apparently formidable problem of explaining them in terms of chemical constitution as modified by varying the pH, with quite a fair degree of success'.
As in Part 10 of this series he assumed that all are constitutionally related to a fundamental substance of known absorption. This is bis-p-hydroxyphenylcarbinol (p-p-dihydroxybenzhydrol). (structure 10). He referred to a previous publication where he explained the fundamental absorption of this substance from first principles.20
Taking this compound as the fundamental substance with absorption at λ 539, and assuming the factor for replacement of -OH by -NH2 tobe1.008and the one for replacing -OH by NMe2 to be 1.058, he obtained the following striking results: the calculated λ for aminohydroxybenzhydrol (alkaline) was 543 nm (observed: 541 nm), that for diaminobenzhydrol (acid) 548 nm (observed 548 nm), that for dimethylaminohydroxybenzhydrol (neutral or alkaline) 570 nm (observed: 571 nm), for amino-dimethylaminobenzhydrol (acid) 574.5 nm (observed: 574 nm), and for tetramethyldiaminobenzhydrol (acid) 603 nm (observed: 603 nm).

These are all the possible cases and from these he could pass on to the malachite-green type of triphenylcarbinol dyes. The factor for replacing H in the centre by C6H5 (or Ph) is 1.025, and the following striking calculated results are obtained (observed values are given in brackets):
Benzaurine 552.5 nm (553 nm), aminofuchsone 556.5 nm (558 nm), dimethylaminofuchsone 561.5 nm (562 nm), dimethyl-aminofuchsonium chloride 589 nm (590 nm), malachite green 618 nm (618 nm).
Explaining all of this is that the electron movement causing the colour is (1) the same for a 3-ringed symmetrical substance as for a 2-ringed substance containing the same auxochrome; (2) nearly the same but slightly perturbed in 3-ringed substances with only 2 symmetrical auxochromes; (3) similar but markedly perturbed when the inactive autochromes have markedly different effects from one another (NMe2 as compared with the other 2); and (4) still more dissimilar when the active auxo-chromes are different (unsymmetrical).
An important point is that the electronic movement causing the high colours cannot take place unless there is a balance or competition between two or more positive centres for a single electron. When one of the NH2 or NMe2 groups is fully ionized by means of mineral acid, it behaves almost as if it were totally absent. Thus crystal violet of λ 596 turns green when made acidic: λ 596 disappears and λ 632 appears (Scheme 8).

He compared this change with other examples (iodine-green, p-nitromalachite-green, malachite-green and p-aminomalachite green), showing conclusively that the group-ring-NHMe2Cl fully ionized is merely a loaded benzene ring. He took it further by acidifying the green solution of acidic crystal violet which then turns yellow: l 596 and 632 both disappear and l 417 appears, giving the product shown below. He again showed that the second-ring-NHMe2Cl behaves merely as loaded benzene.

3.2.4. Substitution in Phenolphthalein and Fluorescein
In Part 15 of the series he made a systematic study of fluorescein.39 He referred to previous work (Part 11) where a study of phenolphthalein was carried out, and the colour-value of substitution by Br as standard substituent was calculated for every one of the 12 H atoms in the compound. He now produced the same results in a different form, viz. that of the dicyclic colour-factors explained in Parts 10 and 14. A table followed where substitutions of Br for H in the 12 different positions, designated by letters a to l, were listed with their respective colour-factors. As an example the colour of phenol-tetrabromo-phthalein (which is phenolphthalein with Br for H in the positions a, b, c and d), was given as 554 X 1.018 X 1.002 X 1.0145 X 1.0180 = 583.6 nm, where the factors used were those in the table for the particular position a, b, c or d. Previous work showed that the colour factor for Cl differed only from Br by 1 part in 1500: the calculated colour for phenol-tetrachlorophthalein is 583.6 (the value for tetrabromophthalein) multiplied by (1499/1500)4, since Cl has replaced Br four times, giving 582.1 nm, agreeing with observations.
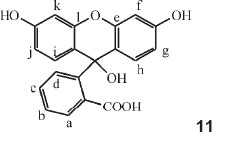
Fluorescein
The derivatives of fluorescein were then examined, employing the same numeration of the positions, shown in structure 11, as was used for phenolphthalein in Part 11 (i.e. assuming that fluorescein is e-l-oxophenolphthalein, it is to be expected that the same colour-factors would hold). Tabulated calculated values were used for any possible substitution product of fluorescein (λ for fluorescein is 493.5 nm) with the observed λ value given. The substituent occupies a particular numerated position within the molecule: e.g. a-monobromo = 1.0162, b-monobromo = 1.000, c-monobromo = 1.0091, etc. Two examples of his calculations follow:
Octobromofluorescein: 493.5 X 1.0162 X 1.000 X 1.0091 X 1.0162 X 1.0122 X 1.0157 X 1.0122 X 1.0157 = 543 nm, with the observed value being 544 nm. Here the Br occupies positions a, b, c, d, f, g, j and k.
Octochlorofluorescein: l for octobromofluorescein X (1499 / 1500)8 = 540.0 nm (observed: 537 nm).
Similar colour factors are used for nitro- and methyl-derivatives.
Part 21 of his series dealt with methyl derivatives of phenol-phthalein.40 Earlier work (Part 11) examined the bromo-derivatives of p-p'-phenolphthalein to investigate the phenomenon of 'loading' in a single-coloured substance. Now methyl derivatives of phenolphthalein were studied and he found that the effect of 'loading' is perfectly regular and additive. He presented a table listing 19 derivatives of phenolphthalein, 2-methyl, 3-methyl, 3,3'-methyl etc., with their respective l values in aqueous alkali. Another table contained the calculated additive values for each load, including data from Part 11, e.g. 3-and 5-methyl = 7.5,3-and5'-methyl = 6.5, etc. From these tables the absorption of any complex halogen or methyl or isopropyl derivative of phenolphthalein can be calculated by adding to the number 554 (l of phenolphthalein in aqueous alkali) in succession to the corresponding figure for each load. Calculations agreed with observed values within experimental error. Similar investigations were presented with o-p-phenolphthalein.
3.2.5. Organic Fluorescence
Part 19 of the series dealt with organic fluorescence.41 He explored the structural characteristics necessary for organic compounds to fluoresce. Regarding the connection between aromatic rings and fluorescence, no substance fluoresces unless it contains an aromatic ring. However the majority of substances with an aromatic ring do not fluoresce, although practically all absorb UV light and do so with sharp absorption bands. Benzene itself does not fluoresce, nor does phenol, but a benzene solution of quinol has a violet fluorescence in strong light. The other two isomers of quinol do not fluoresce appreciably, whence it follows that in addition to an aromatic ring there is a second factor required.
1. This consists of the nature and arrangement of the groups attached to the ring. Regarding the nature of such groups it is apparently necessary that one of these shall be an auxochrome (defined in Part 17 as OH, NH2, NHAlk and NHAlk2) when only one aromatic ring is present; but if two aromatic rings are present no auxochrome is necessary and fluorescence appears whenever two aromatic rings are joined at two places by two groups of almost any sort.
2. The arrangement of the groups attached to the aromatic ring (when there is only one) is probably less important than their nature: bright fluorescence goes with the ortho and para positions, but faint fluorescence is known for substances having their groups in the meta position (e.g. m-nitro-dimethylaniline).
In his paper he presented various categories of fluorescence: amongst the naphthols in aqueous alkali. It was found that two compounds fluoresce (structures 1 and 2), and four do not (sodium phenoxide, alkaline o-cresol, m-cresol, p-cresol). On the other hand, alkaline sodium salicylate (structure 3) fluoresces markedly.
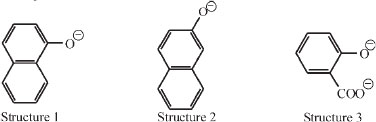
Also all anthranilic acid derivatives in every medium fluoresce: structure 4 in acid, neutral or alkaline, and structure 5 as the ester.
It is easy from these examples to say what is NOT the cause of fluorescence, e.g. (1) the co-operation of two ortho groups or (2) the formation of a side ring between two ortho groups. In the compounds with two ortho groups it is apparently necessary for fluorescence that one group shall be an auxochrome and that the other shall contain an unsaturated group attached to the element nearest the ring. For him it was however difficult to see why o-nitrophenol does not fluoresce unless it has a different formula which is given, and it was also hard to see why it is only the ortho (and not the para and meta) arrangement of the auxochrome and unsaturated group that goes with fluorescence. He suggested that the reason was spatial.
The derivatives of quinol (hydroquinone, p-dihydroxybenzene) do indeed possess fluorescence as striking as that of fluoresceine and rhodamine in spite of their relatively simple constitution. He suggested that is due to the co-operation of two pairs of factors which are the same as in sodium salicylate (structure 3).
Fluorescent substances containing two aromatic rings show fluorescence without any auxochrome; it is only necessary that the rings be doubly united. Some of the 14 compounds he investigated which fluoresce in concentrated sulphuric acid are shown below; others are dihydroanthracene, dihydrophenazine, phenoxazine, xanthhydrol, anthracene, acridine, phenazine, phenoxazonium salts, anthraquinone, acridone and phena-zine-oxide.

It is not possible to assign a quinonoid structure to the solution of xanthenes in sulphuric acid and it appeared quite clear to him that the fluorescence depends not on unsaturation outside the ring, but wholly on the rings as such, with the two groups holding them in rigid position, i.e. on purely spatial considerations.
He concluded by saying that fluorescence is merely the reflection of light from such rigid molecules as are devoid of rigidity,
i.e.  is not fluorescent. As the'mirrors'are much smaller than the waves of light, the reflection is affected by diffraction phenomena, giving a change in colour.
is not fluorescent. As the'mirrors'are much smaller than the waves of light, the reflection is affected by diffraction phenomena, giving a change in colour.
3.2.6. Doubly-linked Diphenylene Compounds and their Halochromic Colours
Halochromism was a term used to describe the colour change or displacement of the visible absorption band of a substance accompanying a change in pH of the solution.
In Part 25 of the series he investigated diphenylene compounds linked by different elements, O and O, S and O, Se and O, and Te and O.42
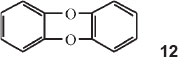
He made diphenylene dioxide, structure 12 above, containing two benzene rings linked in the two ortho positions by two oxygen atoms. Contrary to all expectations this substance gives a blue sulphuric acid solution, although colourless in all other solvents. He then made all possible compounds in which two benzene rings are connected in the ortho position by two identical or different elements or their oxidized derivatives. He presented a long table of the various compounds giving the grouping, the λ's in sulphuric acid and the wave numbers of those absorptions. He then showed that the wave numbers of the strongest lines can be made identical by dividing by different integers. Thus the wavenumber of phenoxine (diphenylene dioxide) is 14650 cm-1 (one of them): this divided by 6 gives 2442 cm-1; the wavenumber for phenthioxine is 17070 cm-1 and that of phenoxselenine is 17150 cm-1. Dividing each by 7, one obtains 2439 cm-1 and 2450 cm-1, respectively. Taking the wavenumber of phenoxtellurine (19530 cm-1) and dividing by 8 one gets again 2441 cm-1. He drew the following conclusions:
a) The colours have very little to do with the actual linking elements, but depend on the fact that all the molecules are of the same shape and are very nearly all of the same size. The actual wide differences in the positions of the absorptions are really due to change in the harmonic number or of some similar integral constant in wave-mechanics theory.
b) The above applies to 'hexad' elements, but can be made applicable by further division sums to all the observations. If the value 2440 (waves per cm) is divided by 4 and then allowed to vary very slightly according to the chemical constitution of the substances between the limits of 594 for the lowest element and 612 for the highest, all observations can be represented in the form of this 'constant' multiplied by a couple of integers.
3.2.7. Colour of Flowers
James Moir was fascinated by the pigments of flowers and published three articles dealing with this under the topic of colour and chemical constitution.22,43,44 A.G. Perkin, the famous dye chemist, had completed his classic investigation of the yellow pigments occurring in petals and isolated nine compounds all having the same chemical skeleton, but differing from one another in the number of hydroxyl groups attached to that skeleton.43 This number is a maximum of seven in the highest member and nil in the parent substance or skeleton flavone or 2-phenylbenzopyrone.
Most of the pigments have hydroxyls in the 5- and 7-positions. Later Willstaetter studied the pigments of higher colour, i.e. scarlet, rose, purple and blue shades, and found all of them to be hydroxyl derivatives of a parent skeleton, known as flavylium chloride or 2-phenylbenzopyrylium chloride.43
Moir realized that the yellow (or flavone) pigments are the oxidation products of the blue ones and he 'arrived at the marvellous result that in nearly all cases Nature is using the same chemical proto-skeleton flavene as the foundation of her pigmentary work'.
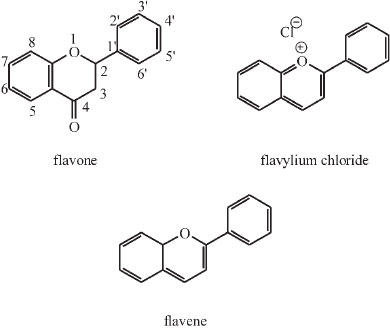
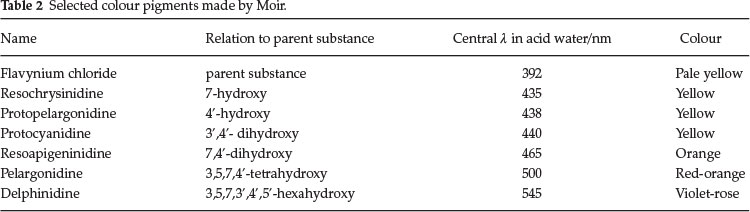
He synthesized many of the members of the series, even those which do not occur in Nature but are theoretically important. Pelargonidine (3,5,7,4'-tetrahydroxyflavylium) occurs in geranium petals as a glucoside and this was synthesized by Willstaetter, the German chemist who won the Nobel prize in 1915 for his work on plant pigments. Later Robinson and Pratt invented a general method whereby all members of the series could be synthesized. Moir used this latter method. He prepared six of the members of the series for the first time, and analyzed the spectra of all of them.
Table 2 sums up some of his results. He concluded that there is a gradual change in shade as the anthocyan is simplified down to its parent substance, as the OH groups are removed one by one, but the removal of hydroxyl from certain positions has much more effect than from other positions, i.e. the 4'- and 7-positions are the most important ones for heightening the colour.
These flower pigments change colour when the solvent changes from acid to neutral and he investigated the colours of them in media of pH 5 and pH 6. In addition he examined the spectrum in alkaline media and proposed that the substances have the configuration of hydrols (carbinols) and are thus analogues to dihydroxybenzhydrol, phenolphthalein and other dicyclic substances.
In some further work he investigated the distance between the auxochrome and the other active centre of the molecule (in this case the oxonium atom, H3O+ part) and found that an increase in the distance caused a striking rise in the 'height' of the colour.
In a further publication he examined the colours of local flowers.43 He applied the knowledge of the spectra of standard pigments (pelargonin, pelargonidin, cyanin, cyanidin, delphinine and delphinidin) to discover what pigment was causing the colour in unknown cases. He proved the presence of glucosides of delphinidin in nearly all bright-blue flowers. Delphinine is rose-coloured as the oxonium salt, and flowers containing it are blue agapanthus, poinsettia and others. He detected glucosides of pelargonidin in Barberton daisy and others. Glucosides of cyanidin were found in many different flowers, amongst which were scarlet zinnia, pink and blue hydrangea and others.
He established that every stage in the origin of flower-colours can be followed by examining all the intermediate substances in solution in concentrated H2SO4 and gave a table with the precise λ value for each pigment.
In his last paper,44 published posthumously in 1930, he further investigated the pigments of yellow flowers, being members of the flavone family. He synthesized a whole range, starting with flavone and adding hydroxyl groups at various positions to examine the spectrum in concentrated H2SO4, in alkaline solution and in near neutral media. The results appeared to show that the effect of each hydroxyl in raising the colour is much the same until the complexity of the pigment quercetine (containing
5 OH's in positions 3,5,7,3' and 4') is reached and thereafter was small.
4. An Appreciation of James Moir as Organic Chemist
This article shows that James Moir was a great synthetic organic chemist and early spectroscopist, having prepared hundreds of highly complex organic compounds, in order to examine their spectra, with the limited instrumentation of his day. His immense study of the colour and constitution of organic compounds is impressive, if one thinks that this work was carried out 100 years ago. He was an expert on phthaleins and his numerical solution to predict the band centre λ of these complex compounds is remarkable. During his organic chemistry research work he covered many topics ranging from dyes and organic fluorescence to colouring pigments in flowers and unusual organic compounds like doubly-linked diphenylenes.
Obviously, his theory of colour was limited in the light of today's understanding of the absorption of light as arising due to the interaction between the oscillating electric field of a light wave and the changing electric dipole of a vibrating bond. His latest theory was based on Bragg's recent X-ray analysis of benzene and its derivatives,21 where he said that the benzene ring is buckled and not a flat hexagon, which we now know is untrue.11 However in his day, he was at the forefront of the development of theories dealing with colour.
Consulting a book on the subject entitled Colour and Constitution of Organic Molecules by John Griffiths published in 1976,45 it is astonishing that Moir's name is nowhere mentioned in the lists of references, since he did so much in this particular field! It is hoped that this article will lead to an appreciation of the tremen dous body of work that James Moir contributed to this field about 100 years ago.
Acknowledgement
The author would like to thank Dr Benita Barton for her enthusiastic help and advice on some of the organic chemistry content of this article.
References
1 P. Loyson, S. Afr. J. Chem, 2014, 67, 61-66. [ Links ]
2 J. McCrae, Obituary in J. Chem. Soc., 1929, 2973. [ Links ]
3 Dorland's Illustrated Medical Dictionary, 30th edn., Elsevier, Philadelphia, 2003, p. 56. [ Links ]
4 F.R .Japp and J. Moir, J. Chem. Soc., Trans., 1900, 77, 608-645. [ Links ]
5 X.-F. Huang et al. J. Organomet. Chem., 2006, 691, 1065-1074. [ Links ]
6 J. Moir, J. Chem.Soc., Trans., 1902, 81, 100-117. [ Links ]
7 I.L.Finar, Organic Chemistry, vol. I, 4th edn., Longmans, Green and Co, London, 1963. [ Links ]
8 I. Guareschi, Atti R. Accad. Torino, 1892, 28, 330. [ Links ]
9 I. Guareschi, Atti R. Accad. Torino, 1898, 34, 27. [ Links ]
10 I. Guareschi, Chem. Centr. 1899, i, 289. [ Links ]
11 B. Barton, private communication. [ Links ]
12 University of Aberdeen, Archives, Special Collection, GB 0231. [ Links ]
13 J. Moir, J. Chem. Soc., Trans., 1907, 91, 1305-1311. [ Links ]
14 J. Moir, Trans. Roy. Soc. S. Afr., 1910, 2, 205-210. [ Links ]
15 J. Moir, J. Chem. Soc., Trans., 1921,119, 1654-1668. [ Links ]
16 J. Moir, J. Chem. Soc., Trans., 1924,125, 1134-1141. [ Links ]
17 J. Moir, J. Chem. Soc., Trans., 1924,125, 1548-1551. [ Links ]
18 J. Moir, J. Chem. Soc., Trans., 1925,127, 967-972. [ Links ]
19 J. Moir, J. Chem. Soc., Trans., 1925,127, 2338-2343. [ Links ]
20 J. Moir, J. Chem. Soc., Trans., 1927, 129, 1809-1823. [ Links ]
21 SACI Proceedings, June 1925, 22-27. [ Links ]
22 J. Moir, Trans. Roy. Soc. S. Afr., 1928,16, 121-130. [ Links ]
23 J. Moir, Trans. Roy. Soc. S. Afr., 1918, 7, 5-14. [ Links ]
24 J. Moir, Trans. Roy. Soc. S. Afr., 1918, 7, 111-116. [ Links ]
25 J. Moir, Trans. Roy. Soc. S. Afr., 1918, 7, 123-127. [ Links ]
26 W.R. Orndorff and R.R. Murray, J. Amer. Chem. Soc., 1917, 39 (4), 679-697. [ Links ]
27 J. Moir, Trans. Roy. Soc. S. Afr., 1918, 7, 183-188. [ Links ]
28 J. Moir, Trans. Roy. Soc. S. Afr., 1919, 8, 42-44. [ Links ]
29 H.E. Howe and K.S. Gibson, Phys. Rev. 1917, 767. [ Links ]
30 J. Moir, Trans. Roy. Soc. S. Afr., 1919, 8, 225-228. [ Links ]
31 J. Moir, Trans. Roy. Soc. S. Afr., 1919, 8, 303-311. [ Links ]
32 J. Moir, Trans. Roy. Soc. S. Afr., 1921, 9, 129-136. [ Links ]
33 J. Moir, Trans. Roy. Soc. S. Afr., 1921, 9, 205-216. [ Links ]
34 E.R. Watson, Colour in Relation to Chemical Constitution, Longmans, London, 1918. [ Links ]
35 J. Moir, Trans. Roy. Soc. S. Afr., 1922,10, 35-39. [ Links ]
36 J. Moir, Trans. Roy. Soc. S. Afr., 1922,10, 65-74. [ Links ]
37 J. Moir, Trans. Roy. Soc. S. Afr., 1922,10, 273-279. [ Links ]
38 J. Moir, Trans. Roy. Soc. S. Afr., 1928,17, 51-59. [ Links ]
39 J. Moir, Trans. Roy. Soc. S. Afr., 1922,10, 159-164. [ Links ]
40 J. Moir, Trans. Roy. Soc. S. Afr., 1926,14, 233-236. [ Links ]
41 J. Moir, Trans. Roy. Soc. S. Afr., 1925,12, 45-50. [ Links ]
42 J. Moir, Trans. Roy. Soc. S. Afr., 1929,18, 137-142. [ Links ]
43 J. Moir, J. S.Afr. Chem. Inst. 1927,15, 36-46. [ Links ]
44 J. Moir, Trans. Roy. Soc. S. Afr., 1930,18, 183-188. [ Links ]
45 J. Griffiths, Colour and Constitution of Organic Molecules, Academic Press, London, 1976. [ Links ]
Received 2 December 2013
Revised 1 September 2014
Accepted 1 September 2014
* E-mail: peter.loyson@nmmu.ac.za


{kind=link}