










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Chemistry
versão On-line ISSN 1996-840X
versão impressa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban Jan. 2014
REVIEW ARTICLE
James Moir as Inorganic Chemist
Peter Loyson*
Chemistry Department, Nelson Mandela Metropolitan University, P.O. Box 77000, Port Elizabeth, 6031, South Africa
ABSTRACT
James Moir was a pioneering chemist in the early 1900s who played a leading role in various chemical societies in South Africa. Although he was mainly an organic chemist, he also made contributions in the field of inorganic chemistry. His forays in this field deal with gold extraction using the new solvent thiourea, removal of cyanide using ferrous sulphate, and investigations into the 'purple of Cassius'. He was also a theoretical chemist who played a role in the development of atomic theory and made suggestions to help unravel the nature of the atom, the composition of the nucleus and chemical combination.
Keywords: Inorganic chemistry, gold, atomic theory, history of chemistry.
1. Introduction
James Moir (1874-1929) played a leading role in the field of chemistry in South Africa. He arrived in this country from Scotland, where he graduated from the University of Aberdeen with a DSc in 1902. He first acted as a chemist in the laboratory of a gold mining company and in 1904 was appointed chemist to the Transvaal Department of Mines. In 1914 he joined the staff of the Government Analyst at the Government Chemical Laboratories in Johannesburg, a position which he held for 15 years until his death. He was twice President of the South African Chemical Institute (1916, 1925), President of the Chemical, Metallurgical and Mining Society of SA (1910-1911) and President of the SA Association for the Advancement of Science (1919). He was awarded the South African Research Medal in 19191.
He played an important role in the gold mining industry and in the early years of the South African Chemical Institute and throughout his working life published some 140 papers.2 He was mainly an organic chemist.3 His activities as an organic chemist, chemical analyst, and physical chemist have been described in this journal before.4-6 However, he also made valuable contributions to the field of inorganic chemistry, especially dealing with aspects of gold extraction. He was also a theoretical chemist who played an important role in the early days of the development of atomic theory, the structure of the nucleus and chemical combination. This article covers his research contributions in inorganic chemistry as reported chronologically in the local chemistry journals.
2. Thiourea as a New Solvent for Gold
James Moir's first publication in the inorganic chemistry field is reported in the May 1906 issue of The Journal of the Chemical, Metallurgical and Mining Society of South Africa and deals with the extraction of gold.7 He reasoned that, since thiourea is made by heating ammonium thiocyanate (1), it might possibly have a similar solvent action on gold, as cyanide. In fact, it turned out that thiourea, which he called thiocarbamide, was an efficient solvent for gold, and if it were not so expensive, it might have been suitable for technical use.↓

He found that gold, in the form of leaf, dissolved in an acidified solution of thiourea in the course of a few hours without the use of an oxidant. However, in the presence of suitable oxidants, the action was very rapid with the gold leaf dissolving in about 15 seconds once a small quantity of ferric chloride or chromic acid was added.
In leaching trials on gold-containing ores, he found that thiourea acted best in faintly acid solutions. For example 61 % of the gold was extracted from a sand charge in three days of frequent agitation, using no oxidant, by an M/80 (0.0125 M) solution. He suggested that the reagent offered promise for use in re-treatment of residue dumps.
Modern studies have shown that acidic solutions of thiourea offer potential applications for the extraction of gold.8-10 Additional studies showed that acidic thiourea extracted 85 % of the gold and silver from a complex sulfide ore after a pre-leach using ferric chloride.11 The optimum concentration for both the thiourea and H2SO4 used in these trials was 30 g L-1. However, there was an unaccounted consumption of thiourea.
Moir studied the properties of thiourea and found that its most remarkable property was its power to form compounds with many other substances, enabling it to dissolve many of the common insoluble precipitates, forming what he called 'double compounds', i.e. a combination of a neutral salt with thiourea. One example was NH4SCN.CS(NH2)2 which behaved as a single substance with a definite melting point of 145 0C.7 Further examples that he studied were Cu(tu)3Cl, Ag(tu)2Cl and others, where tu represents thiourea. It is of interest that the compound Cu(tu)3Cl has Cu and S atoms bonded in an infinite chain of [Cu(tu)4]+ tetrahedra with two shared S atoms at the corners as shown in Fig. 1.12
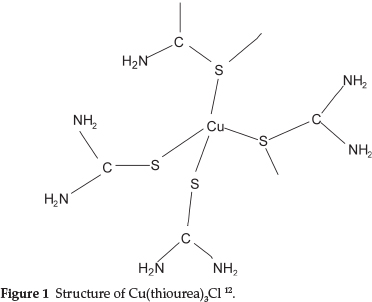
The structures of Ag(tu)2Cl and Au(tu)2Br have also been determined and shown to be similarly complex compounds.13,14
Moir succeeded in isolating two new compounds of gold and thiourea: one which he obtained from gold in a solution of thiourea with sulfuric acid and hydrogen peroxide, which he analyzed as Au2(tu)6SO4 and the other from an HCl medium corresponding approximately to Au3(tu)8Cl3.7 He had difficulty analyzing the correct composition of these compounds: modern analysis shows that gold and thiourea in acid medium form the single complex Au[CS(NH2)2]2+ which should give the salts Au2(tu)4SO4 and Au(tu)2Cl.15,16 His analysis methods were as follows:7
- Au determined by direct ignition, which was known as one of the most accurate processes in analytical chemistry;
- S by oxidation to SO42" and precipitation as BaSO4 and
- Cl by AgCl precipitation after treating the compound with NaOH, which separates out the Au and treating the alkaline filtrate with excess AgNO3 and separating the AgCl from any Ag2S with ammonia.
He bemoaned the absence of any satisfactory apparatus in the country which would permit the determination of H in the compound, as this would be the crucial analysis to confirm the correct composition of these compounds.7
3. Suggestions for a New Atomic Theory
In 1909 he proposed a new theory to suggest probable structures for the known elementary atoms by supposing them all to be built up of different arrangements of four 'primae materies', or elementary building blocks, each with definite properties.17 Using these structures he hoped to explain the selective affinity of the atoms for certain particular atoms in terms of shape and fitting together. He found that the first 20 or so elements could be constructed satisfactorily out of these four elementary building blocks, and the rest were repetitions and imitations according to the Periodic Law.
The fundamental concept of his theory was the nature of the carbon atom. Since all four of its affinities are equal in strength and space-distribution, he represented the carbon atom as made up of four sub-atoms of atomic mass 3.0 and arranged these equidistant in tetrahedral order. He called this sub-atom [C/4], Zoikon, symbol z, atomic mass 3.0.
Secondly he retained hydrogen as an element and building block, since its spectrum showed that it is simple in structure. Thirdly, he postulated a sub-atom x of atomic mass 2.0, monovalent like hydrogen, yet not fully saturating another element when joined to it, and therefore capable of shifting its place within the atom-complex. In addition, he used He as a building block. He maintained that He and Ne were probably true elements, whereas Ar, Kr and Xe were not. Finally, he assumed the definite metals to contain hydrogen as the cause of their electro-posi-tiveness, and that the sub-atom x conferred electro-negative-ness. So his four building blocks were: Zoikon z, sub-atom x, H and He.
With these simple suppositions he could further formulate the elements nitrogen, oxygen and fluorine. As regards nitrogen he could account for (1) its tri- and pentavalent properties, (2) the spatial equivalency of four of the valencies in pentad nitrogen and (3) the fact of the combination NR4 acting as a metal.
The formula he proposed for N was an internal condensation of the carbon tetrahedron with the sub-atom x, which now occupies one of the points of the tetrahedron, as shown in Fig. 3(a), where the crossed circle represents the sub-atom x and the dark circles are again the sub-atom z. The atomic mass works out to 4 x3 + 1x2 = 14 units. The other three points then agreed perfectly in situation with the properties of trivalent N.
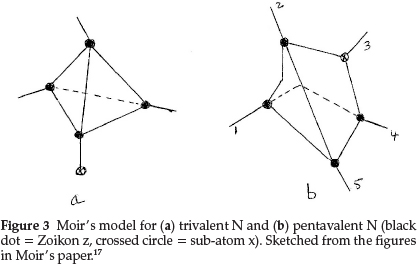
For pentavalent N his model, shown in Fig. 3(b) had the sub-atom x inside the tetrahedron, leaving the four valencies equal and equally distributed as in carbon, but, in addition, conferred an electropositive property on the whole system. In this case the tetrahedral valencies could be satisfied by H or alkyl groups and the combination NR4 or z4xR4 could act as a whole and could simulate an alkali metal.
Regarding oxygen, which requires an increase of 2 units in atomic mass, he suggested two formulae: one to express its ordinary divalent form, and the other for its tetravalent form, which he supposed to be present in salts of pyrone, the oxonium dyes, etc. The former is obtained by repeating for a second time the process giving N from C with constitution z4x2, as shown in Fig. 4(a). This gave an atom with two main valencies inclined to one another at an angle of 109 0, which agreed well with the usual properties of O. The corresponding tetravalent form, shown in Fig. 4(b), with one x in the centre also confirmed the prevailing hypothesis that liquid water was H10O5, consisting of five (H2O) molecules joined up in a regular pentagon, whose angle is 1080, as shown in Fig. 5(a), indicating minimum distortion of the oxygen affinities involved.
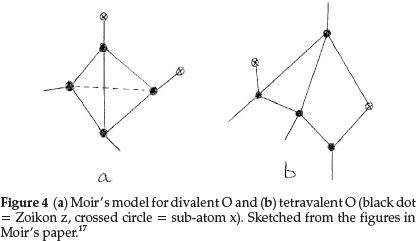
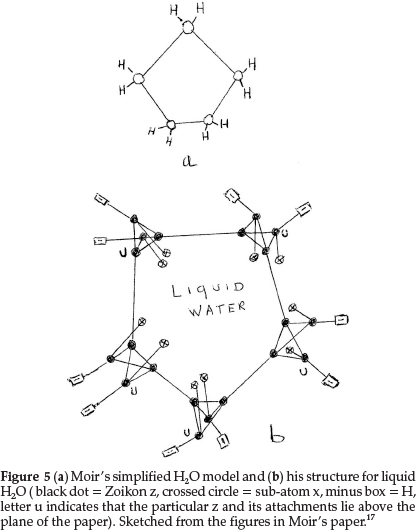
Moir's more detailed structure for water in the liquid state is shown in Fig. 5(b) based on the accepted view in those days.17 It shows five water molecules arranged in a regular pentagon, with each O being tetravalent, with 2 bonds linked to H's (minus boxes) and two other bonds linking each O to neighbouring water molecules. Two sub-atoms x (indicated by a crossed circle) form part of each O structure, as shown in Fig. 4(b).
Some comments can be made at this stage. Firstly H-bonding was not yet discovered: it was first suggested in 1912 by Moore and Winmill,18 to account for trimethylammonium hydroxide in water being a much weaker base than tetramethylammonium hydroxide, due to the special bond being formed between the amine N and the H part of H2O. Only in 1920 did Latimer and Rodebush use this type of bonding to discuss highly associated liquids such as water and HF19
Secondly the structure of liquid water is still an interesting problem. There is no doubt that water, like other liquids, has a structure that involves a great deal of randomness. Yet, it is likely that there are certain configurations of groups of water molecules that occur with high frequency in the liquid.20 Some suggestions are that water retains in part some hydrogen-bonded structure similar to ice, with each O atom being tetrahedrally surrounded by four other O atoms,20 in a three-dimensional network of six-membered rings.21 With limited correct information available at the time, Moir suggested the structure for water as shown in Fig. 5(b), to explain the prevailing idea that liquid water consisted of clusters of five water molecules, held together by O bonds.
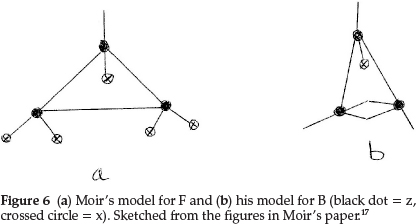
Regarding the element F, Moir proposed a triangular shape, making it 3z with five of the six free bonds satisfied with x's, in order to account for the atomic mass of F being 19.0, giving F = z3x5, as shown in Fig. 6(a). This accumulation of anti-metallic sub-atom x then explained the extraordinary affinity of free fluorine and would explain why HF is capable of condensing to H2F2 at low temperatures.
The next element is neon, which he represented as a tetrahedral arrangement with zx at each of the corners, i.e. Ne = z4x4, giving an atomic mass of 20 units, and this explained its non-valency quite perfectly.
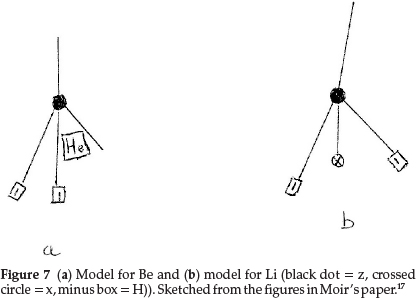
Working back from carbon to complete the first row of the periodic table, boron resembles carbon quite closely and he formulated it as z3x, as shown in Fig. 6(b), giving an atomic mass of 11.0 units. The next two elements, beryllium (which he called glucinum) and lithium, are metals and markedly electro-positive; this property he associated with the presence of hydrogen in the atom; therefore Be was most probably H2zHe, giving an atomic mass of 9.0 units as shown in Fig. 7(a), where the minus squares indicate H's, whilst Li was H2zx with an atomic mass of 7.0, as shown in Fig. 7(b).
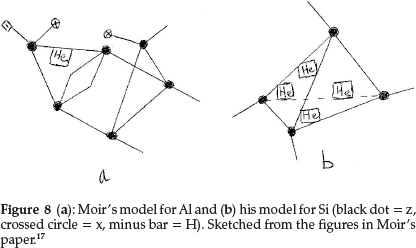
He now used combinations of the first row elements to unravel the structures of the next row. Na differs from Li by exactly 16 in atomic mass, and hence he wrote Na = Liz4x2 = H2z5x3. Mg has a marked fractional atomic mass which he gave as 24.35 and does not resemble Be very closely except in divalency. Assuming He to have an atomic mass of 4.09, he suggested for Mg H2z2He4, giving the required valency of 2, atomic mass and electroposi-tiviness. Al, similar to Be, has 1 He atom with the rest of the skeleton being a duplication of the boron atom, modified by the introduction of hydrogen to account for the metallic character of the element, i.e. Al = Hz6x2He, as shown in Fig. 8(a). Si comes next, represented as the carbon tetrahedron distorted by the insertion of a reversed tetrahedron composed of He atoms, i.e. Si = z4He4, giving an atomic mass of 12 + 16.36 = 28.36, and the model is shown in Fig. 8(b).
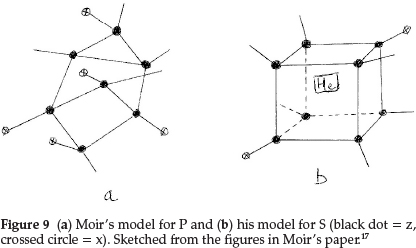
Phosphorus was difficult to represent. It resembled N in valency and its atomic mass difference from N was an abnormal 17.0, and since its atomic mass was a whole number it could not contain He. The atomic mass difference could only be z3x4, with hydrogen being excluded by hypothesis, so that P must be z7x5, giving the structure shown in Fig. 9(a). It is pentavalent with a possibility of trivalency and also explained the electronegativity of the element due to the sub-atom x. Sulfur became z8x2He and its proposed structure is shown in Fig. 9(b). Chlorine, to account for the fraction in its atomic mass, had to contain five He atoms, giving z3x3He5.
He proceeded further building up the rest of the elements, using the same principles, giving a long list of all the remaining elements with their structural formulas. Diagrammatic views of suggested models of some of the elements were projected using lantern slides when he presented this paper at a meeting of the Council in April 1909.
As can be expected, he received support and a fair amount of criticism,22-24 that led to further discussion by Moir.25 It should, however, be remembered that this was in the early days of atomic theory development and according to him it explained the great similarity between C, N and O. It may have given better explanations than the current ones for some of the very remarkable isomeric changes in organic chemistry; for example, the change of ammonium thiocyanate into thiourea could be explained by an intra-atomic movement of the sub-atom x, which he assumed in these formulas. The whole theory was however very speculative and arithmetical and the advancing knowledge of the atom showed a few years later that it had no foundation.3
4. Calculating Atomic Masses
In a subsequent paper from 1909,26 entitled 'The Genesis of the Chemical Elements', he mentioned that he had discovered a new and remarkable relationship between the atomic masses of the elements whereby the accepted values could be calculated with remarkable accuracy. He required the following assumptions: (1) a proton denoted as H* consisting of JJ2 of the atom of hydrogen, (2) another proton called μ consisting of of the mass of an atom of hydrogen and (3) a final proton w consisting of Ί0 of the mass of a hydrogen atom; another assumption he brought in was that the valency of the element depended on the number of times the proton μ occurred in the element in question. He suggested that all the elements were multiples of H* plus the required and regularly varying number of the proton μ; in addition some contained an erratic number of the proton w. He supplied a long table with proposed compositions of the nuclei of the elements, of which the first 15 elements are shown in Table 1.
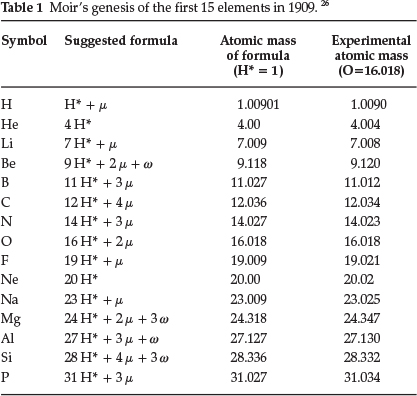
Generally, agreement was very good, for that time. He suggested that the new scheme brought out closer relationships between such groups as the alkali metals and the halogens, and that the Periodic Law may have to be modified from Pt to Bi.
In a later paper from 1912,27 entitled 'Valency and Chemical Affinity', he presented evidence that the proton μ had a mass of about 0.009 atomic mass units, and may be the true cause of valency and chemical combination. He showed this by testing his theory on the experimentally determined molecular mass ratio of KClO3/KCl = 1.643819. Since there are five bonds in KClO3, there are 5 μ' s involved, whilst in KCl there is only one bond, therefore 1 μ, so we have:
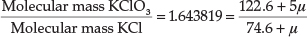
from which μ = 0.00861 mass units. Using the molecular mass ratio of AgNO3/Ag = 1.57473, he obtained a value for μ = 0.008855, but he based his calculation on AgNO3 having eight bonds instead of five, in which case it worked out to about 0.0142 atomic units. Using currently accepted atomic masses for AgNO3 and Ag,28 the ratio is 1.57482, which is surprisingly close to the experimental value he obtained in 1912, but one actually obtains a negative value for μ with this type of reasoning.
5. The Removal of Cyanide
In 1910 he produced a paper entitled 'The Destruction of Cyanide'.29 Since Moir was involved in the chemistry of gold mining he was concerned about the amount of cyanide which accumulated after the gold was extracted and did research so as to minimize its impact on the environment. This paper gave an account of experiments he carried out which contradicted practically everything that was to be found in the prevailing textbooks on the ferrocyanide reaction, i.e. the reaction between ferrous salts and cyanide.29
A preliminary survey of possible methods for destroying cyanides showed that the ferrocyanide reaction was the only one holding out any hope of rapidity of action and reasonable cost. When he and his co-worker Jas Gray, started to investigate the best conditions, they soon discovered that the textbooks 'were at sea' in almost every possible aspect. They found that (1) when excess alkali was used, the amount of ferrocyanide formed was less than when smaller quantities of alkali were present; (2) a rise in temperature above 20 0C was harmful to the reaction; (3) the results were nearly independent of dilution; (4) the reaction was as complete in 5 to 10 seconds as after long standing; and (5) unless the alkalinity be most carefully adjusted to suit the amount of iron used, an excess of ferrous solution gave no better results than the theoretical quantity required by the reaction:

Moir called the precipitate formed 'potassium ferrous ferro-cyanide' and according to Vogel's Qualitative Analysis this white precipitate is formed when ferrous sulphate is added to potassium ferrocyanide.30 However, it rapidly turns blue owing to oxidation. Prussian blue is obtained by mixing Fe3+ with Fe(CN)64- or Fe2+ with Fe(CN)63-, and is represented as Fe4m[Fen(CN)6]3.xH2O,31 and he may have obtained some of this as well when his methods of work-up involved oxidizing agents like KMnO4, peroxide and others.
He maintained that they did not succeed in obtaining entire destruction or removal of the cyanide from solution, although in several cases they obtained a reaction sufficiently complete for industrial purposes, with the cyanide being reduced to 3 or 4 ppm.
Regarding the analytical method used, he pointed out a misconception at the time, namely that when a solution went turbid with one drop of AgNO3, it was therefore free of prussic acid, HCN. This was quite a mistake, since it was merely evidence of the absence of CN-, whereas any amount of HCN could be present and would not be titrated by AgNO3 unless the solution was strongly alkaline. He maintained that many of the Rand cyanide strengths were higher than the reported value to an extent of 10-40 % of the stated value. This was because the titration was not correct, unless the 'protective alkali' greatly exceeded the cyanide present.
However, making the solution strongly alkaline caused changes in the composition of the reaction product. For example, it broke down K2Fe2(CN)6 into K4Fe(CN)6 and Fe(OH)2 and the latter would attack the free cyanide which needed to be determined in solution. They surmounted this problem by two methods: (1) oxidation of the reaction product before rendering alkaline and (2) coagulating the reaction mixture with inert substances like chalk, charcoal and kieselguhr, so as to enable it to be filtered before adding the strong alkali.
During the reading of this paper at Council, some members objected to the use of the word 'destruction', since the cyanide was not destroyed but merely converted to an insoluble form. One should rather try to break the CN bond and the use of alkaline permanganate was suggested.32
6. Colloidal Gold and 'Purple of Cassius'
He carried out a number of experiments,33 to test the generally-accepted view that 'purple of Cassius' was a solid solution of colloidal gold in hydrated stannic oxide. This purple resisted cold pure aqua regia, differing from pure finely divided gold. All experiments were carried out using a very dilute solution of HAuCl4 in water. He found that acid solution of SnCl2 alone produced a pale yellow-brown coloration of remarkable stability, which he investigated using a variety of reagents. To produce the purple of Cassius from HAuCl4and SnCl2, the presence of an oxidant was necessary. Dilute HNO3 had no effect, but heated aqua regia and Cl2 acted at once. Addition of a trace of sodium nitrite gave an intense pink shade of purple of Cassius, but an excess of nitrite prevented the formation of any colour.
Some experiments were carried out to obtain the purple shade without the presence of tin. Hydrazine sulphate gave the same blue-purple shade immediately: he observed that the tin in the purple of Cassius is merely a vehicle for the finely-divided gold and did not enter into chemical combination with it. He also observed that the particle size of gold played an important role in the shade of the colour: large particles producing the purple shade, with even larger ones giving a blue shade and a pink shade produced by gold in its finest state of division. This is generally found to be correct with nanoparticles of gold showing different colours depending on the particle size. Actually, Michael Faraday was the first to recognize that the colour was due to the minute size of the gold particles.34 Nowadays, gold nanoparticle technology plays a key role in many research areas such as electron microscopy, electronics, material science and smart sensors for detecting specific biomolecules and heavy metal ions.35,36
Recent information shows that Purple of Cassius is a purple pigment formed in the reaction of gold with tin(II) chloride.31 It has been used as a chemical test for gold. Generally the preparation involves dissolving the gold in aqua regia, followed by reaction with a solution of stannous chloride. The Sn(II) chloride reduces the chloroauric acid to a colloid of elemental gold, supported on tin dioxide to give a purple coloration, the intensity of the colour being proportional to the concentration of gold. It was supposedly discovered by Andreas Cassius in Leyden in 1685.37
7. Radioactivity and the Periodic Table
To celebrate the 21st year of the discovery of radioactivity by Becquerel in 1896, he presented a paper entitled 'Latter Day Alchemy and Transmutation' on the radioactive elements and new views on the Periodic Law.38 This was one of the first papers published in the Journal of the Chemical Institute, then called the SA Association of Analytical Chemists and was read at a meeting in September 1917. It took stock of the wonderful field of radioactivity and compared spontaneous transmutation of the element radium with what the alchemists attempted in vain to do in the Middle Ages. He described the radioactive disintegration process of U correctly but used strange names for certain of the intermediate nuclides, e.g. Niton (Nt) = Radon Rn; Ionium = Thorium 230; Uranium X = Th 234, and others, ending up with Pb 206. Similarly he described the decay of Thorium ending up with Pb 208. He explained correctly the concept of isotopes and its relation to elements not having an atomic mass equal to an exact integer.
Regarding the Periodic Law, he described the structure of the atom, consisting of a heavy nucleus, with electrons in shells. He warned that the positive charge in the nucleus may not be the only charge in the nucleus, but could be the net charge, for example H may have 5+ and 4- charges instead of +1 charge; no one knew this at that stage.38 He maintained that there still was no satisfactory explanation why oxygen has six valencies, while it appeared to have only two.
The 14 rare earth metals, which are all trivalent, were still as exceptional as ever, and could not be fitted into any periodic table. An ideal table would jump from Ce ( atomic number 58) to Ta (atomic number 73) without any break; however, the X-ray spectrum method showed that the intervening rare elements had the intermediate atomic numbers.
8. The Atomic Theory in 1921
As part of a Presidential Address in July 1921, published in the South African Journal of Science,39 he said that in view of the tremendous upheaval of the foundation of Chemistry and Physics, someone should take the trouble to assimilate the new views and put them on record in a connected form for the use of South African scientists. He maintained that the revolution had reduced Chemistry to a branch of Physics!
This had taken place in three logical steps: (1) the recognition that all chemical properties of a compound could be explained by the number and arrangement of the chemical valencies of the different elements contained in the compound, (2) the Periodic Law of the elements whereby the model atom consisted of a hollow sphere with a distant shell of electrons, their number and arrangement thus explaining all the possible kinds of valencies and therefore all the chemical properties, and (3) the remaining (non-chemical) properties of the atom such as mass which depended on the nature of the nucleus, no longer indivisible, as believed in the 19th century, but was itself a compound of smaller bodies in a special arrangement.
He described that the fundamental concepts of the new atomic theory were those connected with hydrogen in its three forms: H+, called hydrion or the fundamental proton, nascent hydrogen H, and H2 gas and he gave examples for each with notations used in those days. Then he tried to explain the existence of lithium hydride, apparently consisting of two positive substances, Li+ and H+, which was impossible due to electrical repulsion, and concluded that hydrogen gas must consist of two H's, such that one positive H, (H+), is joined to a negative (H+)2-, since electrolysis of LiH gave Li metal at the cathode, and H2 at the anode. He was not aware at that time of the existence of the H- anion in LiH, which would account for the liberation of H2 gas at the anode.
He next described disintegration experiments resulting from bombardments of nuclei with a-particles.39 Rutherford had shown that, by bombarding N2 gas with a-particles, hydrogen nuclei were expelled. Moir therefore concluded that the N nuclei contained hydrogen nuclei, and by magnetic deviation they were shown to have a mass of 1 atomic mass unit and charge +1. Rutherford announced after his experiment that the atomic mass of 14 belonging to nitrogen probably consisted of two hydrogen atoms combined with three helium atoms. On continuing his experiments, however, Rutherford soon found that the H particles were accompanied by another set of characteristic particles 5 to 10 times as many in number, which differed from any known particles. From the range and magnetic deviation they were shown to possess mass 3 and charge 2 and thus were similar to the a-particle with only > of the latter's mass. N of atomic mass 14 was thus inferred to consist of 4 particles of mass 3 instead of three particles of mass 4 in addition to the two hydrogen nuclei.
Many years ago Moir had predicted that N would be found to be Cx, in which x was a sub-atom of atomic mass 2.17 He predicted that C would be found to consist of a tetrahedral arrangement of four sub-atoms of mass 3 and also that an element would exist of mass 3 from N, which he had called Zoikon. Regarding the monovalent element x of two atomic mass units, predicted by him in 1909, Rutherford's experiments seemed to indicate that it only existed in nuclei and that it consisted of (H+e-H+), where e- represents the electron, so that it yielded H+ on impact. He further stated that the a-particle was composed of two of these x elements, and the 3++ particle was composed of one x and an H+. Rutherford's conception of the O nucleus as CHe was the same as Moir's idea of it as Cx2.
In connection with the sub-atom x+, another pioneer of atomic structure, WD. Harkins from the USA, came to the conclusion that it was 'the primary group in atom building', and agreed with Moir that 'the a-particle was x2+', although he used different symbols.40
He then proceeded with comments on the recently published octet theory of Lewis,41 which was almost certainly true for the elements higher than oxygen. F-, Ne and Na+ all had the same octet of electrons, but in the F atom, one was missing, leaving a 'hole'. In Na one was in excess. In NaF the extra electron of the Na filled the hole in the F atom, both becoming ions even in the crystal state and both had completed octets. He said that there were great difficulties accepting Lewis's octet valency theory for the lower elements. His new suggestion was that the arrangement or structure of the nucleus governed the structure of the valency electrons in the smaller atoms. The nucleus thus indirectly influenced the direction in space of the valency electrons and consequently affected the chemical properties of the atom by its arrangement, not by its size. So he maintained that the C-nucleus had the shape of a regular tetrahedron, the N-nucleus had a tetrahedron or pyramid with one point further from the centre than the other three, and the O-nucleus had the shape of an irregular tetrahedron with two points nearer and two points further from the centre. Each valency electron in the case of C had its average position in line with a projection of the nucleus, giving a regular tetrahedral result.
So at that time Moir was still clinging onto his incorrect atomic theory and sought support from Rutherford's experiments, which seemed to confirm some of his suggestions, and from Harkins, another noted figure in the development of atomic theory, who agreed with Moir's conception of the a-particle.
9. Chemical Combination
In 1924 he published an article describing the latest advances in the theory of chemical combination.42 In it he finally admitted that his previous theory described in 1909 was out of date and recanted publically for it, saying science had advanced. Chemical combination or bonding depended entirely on the surfaces of the atoms, in other words, on the outer electrons of each atom. The fundamental concept of chemical combination was that in inorganic chemistry the combination consisted in a metal undergoing ionization, involving the loss of one or more of the valency-electrons existing in its outermost shell; ionization of a non-metal consisted in it gaining one or more electrons which were added to the outermost shell.
Another important point in bonding was the variable valency: he looked at S in H2S and in H2SO4, and said that ionization in H2SO4 was brought about by losing six electrons whilst in H2S it gained two electrons, forming what he called 'sulphidion', the S2- ion. He also mentioned variable valencies in Ti, V Cr and other transition metals.
Another factor in chemical combination was the size of the atoms. He observed that SF6 existed but not SCl6. In the latter case the outer electrons of the six Cl atoms would be brought too close together and would repel one another, whereas the outsides of the six F atoms would not be so close together.
He maintained that the structure of diamond was tetrahedral like in CH4, and that of graphite was the same as that of diamond but loosened out and twisted.
10. Conclusion and an Appraisal of Moir as an Inorganic Chemist
This article has shown that James Moir was an accomplished inorganic and theoretical chemist. His activities dealt mainly with the chemistry of gold and associated aspects, as he was very involved in the gold mining industry. He also played an important role in the early development of atomic theory and chemical bonding and was intimately involved with new developments taking place in the field of inorganic chemistry.
Looking critically at his inorganic chemistry accomplishments it should be mentioned that he was the first to suggest thiourea as a solvent for gold, and his work dealing with the properties of thiourea, leading to the formation of the so-called 'double compounds' was new at the time. Unfortunately he got the analysis of Au2(tu)4SO4 and Au(tu)2Cl wrong, probably due to his lengthy and out-dated methods of analysis and complexity of the structure of these salts. Regarding his work dealing with cyanide removal using ferrous sulfate, he uncovered a lot of new information, which contradicted that found in the prevailing textbooks of the time, and managed to reduce the concentration of cyanide in solutions to a few ppm. He also pointed out a mistake in the accepted analysis of cyanide due to the presence of HCN in addition to CN- and solved this by carrying out the titration in an alkaline medium. Concerning his work on the purple of Cassius, he studied the experimental factors leading to its formation and observed the dependence of colour on particle size of the gold.
His contributions to the development of atomic theory and chemical bonding were, at the time, highly regarded and he was at the forefront of these activities. According to his theory he proposed that all the elements were made up of four building blocks: a sub-atom called Zoikon, z, another sub-atom x, He and H. Using this theory he suggested structures for each element which could explain many of its properties. It also emphasized the great similarity between C, N and O. Unfortunately, his proposed theory later became obsolete, and he publically recanted it in 1924, saying that science had advanced. At the time it was a good theory, however, which according to him explained numerous chemical observations. Even other pioneers of atomic structure, like Rutherford and Harkins, had similar ideas to those of Moir. He also commented on the new Lewis theory, and suggested possible improvements.
Examining his international standing, he published a number of articles in the Journal of the Chemical Society, and especially in its Transactions. Most of these dealt with his activities as an organic chemist and his work on colour of organic compounds.4 Two papers published in the Transactions, however, deal with his inorganic activities: one on thiourea as a solvent for gold,43 and the other on harmonizing atomic weights.44 This indicates that his name must have been known in the international community. This is further indicated by his obituary in the Journal of the Chemical Society in 1929.3 So, he was a chemist of international standing, better known as an organic than an inorganic chemist. He, however, published most of his inorganic research in the local chemistry journals, especially Transactions of the Royal Society of South Africa, and the Journal of the Chemical, Metallurgical and Mining Society of South Africa, which may indicate that his work, especially dealing with his atomic theory, was not that well-known internationally. There is no doubt, however, that his impact on the South African chemical fraternity was massive, as shown by the James Moir Medal which is presented annually to the best chemistry student at Universities in South Africa. He was a very good all-round chemist who called himself a 'chemical investigator'45 and hence was interested in all aspects of chemistry, including inorganic chemistry, which this article has highlighted. The article also gives a good insight into the development of inorganic chemistry over the years 1906-1924.
References
1 SACI Proceedings 1928-1929, September 1929, pp. 29-30. [ Links ]
2 P. Bloom, SACI pamphlet: James Moir Medal Information. [ Links ]
3 J. McCrae, Obituary in J. Chem. Soc., 1929, 2973. [ Links ]
4 P. Loyson, S. Afr. J. Chem., 2014, 67, 124-136. [ Links ]
5 P. Loyson, S. Afr. J. Chem., 2014, 67, 61-66. [ Links ]
6 P. Loyson, S. Afr. J. Chem., 2014, 67, 137-142. [ Links ]
7 J. Moir, J. Chem. Metal. Min. Soc. S. Afr., 1906, 6, 332-336. [ Links ]
8 T. Groenewald, Hydrometall., 1976,1, 277-290. [ Links ]
9 T. Groenewald, J. S. Afr. Inst. Min. and Metall., 1977, 77, 217-223. [ Links ]
10 C.K. Chen, T.N. Lung and C.C. Wan, Hydrometall. 1980, 5, 207-212. [ Links ]
11 R.G. Sandberg and J.L. Huiatt, Recovery ofSilver, Gold and Lead from a Complex Sulfide ore using Ferric Chloride, Thiourea and Brine Leach Solution, U.S. Dept. of the Interior, RI 9022, 1986. [ Links ]
12 G. Nickless, ed., Inorganic Sulphur Chemistry, Elsevier Publishing Co, New York, USA, 1968, p. 61. [ Links ]
13 E.A. Vizzini and E.L. Amma, J. Am. Chem. Soc., 1966, 88, 2872-2873. [ Links ]
14 L.C. Porter, J.P. Fackler, J.A. Costamagna and R.Schmidt, Acta Crystall. C, 1992,48, 1751-1754. [ Links ]
15 J. Marsden and C. I. House, The Chemistry of Gold Extraction, SME, Littleton, Colorado, USA, 2006, p. 335. [ Links ]
16 R. Mensah-Biney, Adsorption of Acidic Gold-Thiourea Complex onto a Strong Cation Exchange Resin, North Carolina State University MRL, Asheville, NC. October 1996. [ Links ]
17 J. Moir, J. Chem. Metal. Min. Soc. S. Afr., 1909, 9, 11, 334-340. [ Links ]
18 T.S. Moore and T.F. Winmill, J. Chem. Soc., 1912,101, 1635. [ Links ]
19 WM. Latimer and WH. Rodebush, J. Am. Chem. Soc., 1920, 42, 1419-1433. [ Links ]
20 L. Pauling, The Nature of the Chemical Bond, 3rd edn., Cornell University Press, New York, USA, 1963, pp. 464-473. [ Links ]
21 B.M. Mahan and R.J. Myers, University Chemistry, 4th edn.,The Benjamin /Cummings Publishing Company, Menlo Park, California, 1987, p.680. [ Links ]
22 J.A. Wilkinson and F.W. Watson, J. Chem. Metal. Min. Soc. S. Afr., 1909, 9, 11, 341-343. [ Links ]
23 P.R. Roux, J. Chem. Metal. Min. Soc .S. Afr., 1909, 9, 392-393. [ Links ]
24 A.Adair, J. Chem. Metal. Min. Soc .S. Afr., 1909, 9, 433-434. [ Links ]
25 J. Moir, J. Chem. Metal. Min. Soc .S. Afr., 1909, 10, 96-99. [ Links ]
26 J. Moir, Trans. Roy. Soc. S. Afr., 1910,1, 413-415. [ Links ]
27 J. Moir, Trans. Roy. Soc. S. Afr., 1912, 3, 1, 39-40. [ Links ]
28 J. McMurray and R.C. Fay, Chemistry, 3rd edn., Prentice Hall, Upper Saddle River, New Jersey, 2001. [ Links ]
29 J. Moir and J. Gray, J. Chem. Metal. Min. Soc. S. Afr., 1910,10,433-441. [ Links ]
30 A.I. Vogel, A Textbook of Macro and Semimicro Qualitative Inorganic Analysis, 4th edn., Longmans, 1968, p. 261. [ Links ]
31 F.A.Cotton and G.Wilkinson, Advanced Inorganic Chemistry, 5th edn., John Wiley and Sons, New York, 1988, p. 721. [ Links ]
32 H.A.White, J. Chem. Metal. Min. Soc. S. Afr.,1910,10, 442-449. [ Links ]
33 J. Moir, Trans. Roy. Soc. S. Afr., 1910, 2, 203-204. [ Links ]
34 M. Faraday, Phil. Trans. Roy. Soc., 1857,147, 145-181. [ Links ]
35 C.N. Ramachandra Rao, G.U. Kulkarni, P.J. Thomas and P. Edwards, Chem. Soc. Rev., 2000, 29, 27-35. [ Links ]
36 S. Zeng, K.T. Yong, I. Roy, X.Q. Dinnh, X. Yu and F. Luan, Plasmonics, 6, 491-506. [ Links ]
37 L.B. Hunt, Gold Bulletin, 1976, 9, 4, 134-139. [ Links ]
38 J. Moir, S. Afr. Assoc. Anal. Chem, 1918,1, 12-18. [ Links ]
39 J. Moir, S. Afr. J. Sc., 1921,17, 6, 47-62. [ Links ]
40 W.D. Harkins, Nature, 1921,107, 202-203. [ Links ]
41 G.Lewis, J. Am. Chem. Soc., 1916, 38, 4, 762-785. [ Links ]
42 J. Moir, J. Chem. Metal. Min. Soc. S. Afr., 1924, 24, 12, 158-166. [ Links ]
43 J. Moir, J. Chem. Soc. Trans., 1906, 89, 1345-1350. [ Links ]
44 J. Moir, J. Chem. Soc. Trans., 1909, 95, 1752-1755. [ Links ]
45 SACI Proceedings, June 1916, pp. 24-31. [ Links ]
Received 28 August 2014
Revised 2 October 2014
Accepted 6 October 2014
* E-mail: ployson@nmmu.ac.za

