










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban Jan. 2014
REVIEW ARTICLE
James Moir (1874-1929) Pioneering Chemical Analyst in South Africa in the Early 1900s
Peter Loyson*
Chemistry Department, Nelson Mandela Metropolitan University, P.O. Box 77000, Port Elizabeth, 6031 South Africa
ABSTRACT
James Moir, a pioneering chemist in South Africa, played a leading role in the SA Chemical Institute. He was an excellent organic chemist, who published widely on the relationship between the colour of an organic substance and its chemical structure. In addition to this, he worked as an analyst in the Government Chemical Laboratories in Johannesburg, where he was exposed to a wide variety of chemical analyses. He developed many new methods of analysis and improved others, which he published in the official journal of the Chemical Institute. This article traces his chemical analysis research work as gleaned from the pages of this journal. It deals with coal analysis, where he developed a simple formula to predict the calorific value, determination of nitrous fumes in air with special reference to fuse-igniters, improving the structure of methyl orange as an indicator, analysis of metals such as Cu, Co, As and Cr, and cyanide analysis. Furthermore, the article discusses his sensitive test for phenol and gives a case study where he did some chemical detective work. From all of this, it is apparent that James Moir was a very fine analyst. He is commemorated by Chemistry students for the 'James Moir Medal', awarded annually to the top BSc (Hons) or BTech student at South African universities.
Keywords: Analysis, early 1900s, education, pioneering chemist, SACI.
1. Introduction
James Moir (Fig. 1) arrived in South Africa from the University of Aberdeen in Scotland in 1902. He had graduated with MA and BSc degrees in 1897 and had just completed his DSc when, for health reasons, he sought the sunny climate of South Africa. After a brief spell as science master at a Johannesburg school, he acted as a chemist in the laboratory of a gold mining company and in 1904 was appointed chemist to the Transvaal Department of Mines. In 1914 he joined the staff of the Government Analyst at the Government Chemical Laboratories in Johannesburg, a position which he held for fifteen years until his death. He was twice President of the South African Chemical Institute (1916, 1925), and also served terms as President of the Chemical, Metallurgical and Mining Society of SA. (1910-1911) and President of the SA Association for the Advancement of Science (1919). He was awarded the South African Research Medal in 1919.1 He played an important role in the gold mining industry and in the early years of the South African Chemical Institute and throughout his working life published some 140 papers.2He was involved in many chemical investigations and was at the forefront of the development of chemistry in South Africa, especially in the field of organic chemistry, where he investigated the connection between the colour of organic compounds and their chemical constitution.3 However, he also made valuable contributions as a chemical analyst and a survey of his research publications in this field shows his wide expertise and keen sense of investigation. This article traces his research output in the field of chemical analysis as reported in the pages of the Journal of the South African Association of Analytical Chemists, which in 1922 became the Journal of the South African Chemical Institute, before it eventually became the South African Journal ofChemistry.

2. Analysing Coal
In 1916 the South African Chemical Institute was still known as the South African Association of Analytical Chemists. When he was elected President in that year he stated in his Presidential Address that he was very honoured to be elected President, because "...he was not primarily an Analytical Chemist, since he was trained almost solely as a chemical investigator."4 In the same address he bemoaned the fact that analysts expressed all their results to two decimal places, regardless of the nature of the material analyzed or the defects of the analytical process. He likened the accuracy of an analytical investigation to a chain, in which the weakest link will decide the value of the whole process. Thus, in testing coal for calorific value by the bomb method, the weakest link was the reading of the temperature difference, which could not be read more accurately than one part in 300 (0.01 °C in 3 °C). Consequently, no other part of the investigation needs to aim at a higher degree of accuracy and it was therefore useless to weigh the coal sample to four decimal places!
Concerning the analysis of coal,5 he developed a formula for calculating the dry-basis calorific value from the % ash on a sample dried at 100 °C. The formula is given by the equation:

He compared the calculated value for a whole range of coal samples with the experimentally obtained value using the Mahler-bomb method and the agreement was remarkable, no matter what the ash and volatile matter were. From these observations he inferred that the carbonaceous part of coal has a fairly constant composition but that it varies in hydrogen and oxygen content in such a way that the calorific value per unit weight remains constant. For example, with bituminous coal represented by C16H16 O2, he suggested anthracites to be represented by C16H8 or C16H4. When a bituminous coal is burned, the heat generated from C16 and H12 is diminished by the cooling effect caused by boiling off 2H2O. The net result may easily be the same as in burning anthracite, C16 and H4, without deduction. He also suggested that coals which deviate from the calculated formula have one more oxygen atom, e.g. C16H16O3 (bituminous) or C16H4O (anthracitic).
3. Determining Nitrous Fumes in Air, with Special Reference to Fuse Igniters
The determination of small amounts (less than 0.10 % by volume) of NO2 in air was a challenging analytical test in those days.6 One such method was the official heat test paper used for explosives. This was a starch KI paper moistened with a 50 % glycerine solution. A brown line developed with time proportional to the concentration of NO2 in the air. He improved this method to make it 15 times more sensitive by substituting water for glycerine. Other methods of dealing with higher concentrations of NO2 depended on oxidizing nitrite to nitrate, using either KMnO4 or H2O2, and determining the nitrate produced. He developed a new method for determining nitrites or nitrous acid which could be used in water analysis.6 It was based on diazotization of the sample, containing say 0.02 g of NO2 gas or 0.015 g of NaNO2 at 50 °C with p-nitroaniline, forming p-nitrobenzenediazonium chloride. A reagent made up of an alkaline solution of a-naphthol in sodium acetate was then added leading to an orange-red precipitate which he called p-nitrobenzeneazo-a-naphthol, which was filtered off on a Gooch crucible and weighed: 293 parts of this dye resulted from 92 parts of NO2 gas or 69 parts of NaNO2. The reaction sequence is shown in Scheme 1.

For minute traces of nitrite one could dissolve the dye in NaOH giving an intense blue-violet colour, so that colori-metric comparison with known nitrite solutions could be carried out.
The analysis of NO2 resulting from blasting in mines was a challenging analytical problem in those days. The miner's fuse-igniter was a long strip of gelatin wound round a stick. This gave off so much NO2 on burning that the government prohibited its use and made a regulation that fuse-igniters must be such that the NO2 given off on burning shall not exceed 0.5 %. Chemical testing required the analysis of a quantity of NO2 (ranging from 2 to 7 mg) distributed over say 12 litres of air and accompanied by smoke which may contain nitrite, nitrate, carbonate and CO2. He developed a method to determine NO2 gas in the presence of nitrite ion,6 which was superior to the alternative method using H2O2 oxidation, and investigated the reaction sequence which the NO2 undergoes. The fumes were treated with concentrated neutral KI solution and agitated for six hours, after which the liberated I2 was titrated against standard 0.100 M thiosulphate, giving experimentally 1.00 cm3 = 0.0051 g NO2, instead of the theoretical 1.00 cm3 = 0.0046 g NO2. In neutral solution the reaction is:

If a trace of acid is present the reaction goes differently and is interminable:
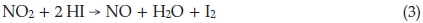
and the NO re-oxidizes to NO2 and repeats the process. A side reaction resulting from the following cyclic process also takes place:
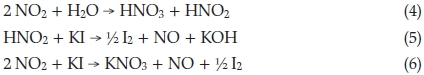
(obtained by adding (4) and (5)).
The NO produced in this side reaction is oxidized to NO2 and by cancelling one obtains the overall side reaction:
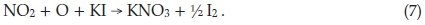
If air containing NO2 is bubbled through KI solution, only half this amount of I2 is liberated, because the reaction is then at the intermediate stage represented by (5), with the NO being carried away and re-oxidized outside the solution.
4. Indicator Work
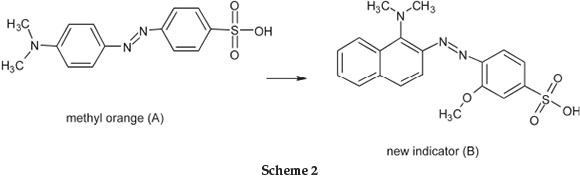
James Moir developed an improved methyl orange indicator by modifying its chemical structure.7 He called methyl orange p-sulphobenzeneazo-dimethylaniline (A). Its limitation was that its change in colour was much less in artificial light than in daylight. The suggested new indicator, which he named p-sulpho-o-methoxybenzeneazodimethylalphanaphthylamine (B), is sufficiently soluble in water, with a change from orange to blue violet in daylight and a range of pH change from 3.5-4.9, whereas that for methyl orange is from 2.8-4.1. Scheme 2 shows the change in structure.
He also developed a polychromatic indicator which he named catecholsulphobenzeine or 3-3'dioxybenzaurine-2'-sulphonic acid, with seven distinct shades in passing from acid to alkali, as shown in Table 1.8 The new indicator belongs to the 'phenol red' class. He made this by condensing catechol with o-benzene-sulphonic acid, and it has four ionizable hydrogens.
5. Copper Analysis
One of the very first papers to be published in the Journal of the South African Association of Analytical Chemists in 1918 was Moir's work dealing with a new volumetric method for the estimation of copper.9 He first introduced a new way of bringing the metal or its ore into a condition for determining the copper present, by dissolving the sample in concentrated HNO3, removing NO by heating, and adding urea, which destroys, upon boiling, any HNO2, which is used up in decomposing the urea into CO2 and N2. The solution was now neutralized short of precipitation, and excess sodium acetate added, which converted the copper into cupric acetate, ready to be analyzed by the new volumetric method consisting of adding a known volume of excess standard thiosulphate, followed immediately by a thiocyanate to precipitate the copper. The cuprous thiocyanate precipitate was filtered off and rejected and the colourless filtrate was diluted, acidified with H2SO4 and the excess thiosulphate titrated with standard I2 using starch as indicator. The HCNS also present is not affected by iodine. The calculation is merely that each cm3 difference between 0.100 N thiosulphate used initially and 0.100 N iodine used in the titration is equivalent to 0.00636 g Cu. The reaction is given below:
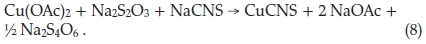
6. Co Determination
Regarding the detection of Co in cobalt-steels,10 he developed a simple spectrophotometric method, by dissolving a suitable sample of the steel in aqua regia and examining the dull yellow solution with the spectroscope, focusing on the extreme red end of the spectrum. If the steel contained 5 % or more of Co, the red end of the spectrum was cut off entirely and two very narrow absorption bands at l 621 nm and l 607 nm were visible in the orange area. With less Co present these two bands in the orange area were not seen but towards the red end of the spectrum a very strong band appeared at l 700 nm and a lesser one at l 661 nm. If the steel contained 5 % or more Co, these strong bands could be brought into evidence by diluting the solution with concentrated HCl. Water destroys the characteristic absorption spectrum and must not be used for diluting the acid solution. Using this method 0.0002 g Co, which corresponds to 10 mg of a steel containing 2 % Co, was easily detected when dissolved in 8 cm3 of aqua regia. Even half of this amount can be detected under favourable conditions.
7. Arsenic Analysis
Moir improved a method, originally put forward by a Past President of the Chemical Institute, Dr H.H. Green,11 for the quantitative estimation of moderate amounts of arsenic, by replacing elemental Zn by an electrolytic H2 generator.12 The method was based on the formation of AsH3 in a stream of H2, and bubbling the AsH3 formed through two successive tubes containing dilute AgNO3 solution. Oxidation of the AsH3 takes place from -3 to + 3bythe Ag+, which is reduced to Ag and the As3+ formed is subsequently titrated with standard I2, using starch as indicator: 1.00 cm3 of 0.00192 NI2 = 0.000100 g of As2O3. He found that As present in the +5 state is reduced to AsH3 just as fast as if it were in the +3 state. The reactions are:
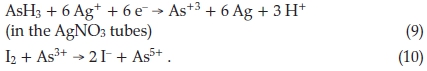
His electrolytic cell for generating H2 is shown in Fig. 2 and was actually an early divided cell, where the two compartments, each containing an electrode, were separated from each other using a porous partition. An inner cylindrical open-ended glass tube contained the cathode, which was a large triangular Cd plate suspended from a hook and connected to the negative pole of a battery; the bottom open end of the cylinder was joined to an alundum porous crucible using a wide rubber tube; a Pt anode was made to fit closely to the outside of the cylinder and connected to the + pole of the battery. The rest of the apparatus consisted of a high narrow beaker to hold the anodic fluid, which was 1:5 H2SO4; this needed to fill the space between the beaker and the cylinder in order for the fluid to exert considerable pressure on the alundum crucible and thus prevent the outward diffusion of the arsenic-containing liquid inside. A thistle-headed funnel was attached to the top end of the cylinder, and a cross tube was attached to this, filled with lead acetated cotton wool, to stop any H2S from coming through. When the apparatus was in use, the whole was immersed in a large tank of cold water.
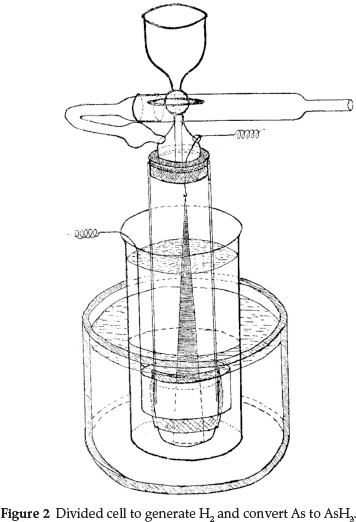
Before use the surface of the Cd was activated by reversing the current and thus making Cd the anode in dilute acid for half a minute, and then restoring the normal current direction until Cd was redeposited in a porous form on the solid Cd. The analysis was started by running in about 12 cm3 of 20 % H2SO4 through the funnel; electrical connections were made and the gas exit was connected to two tubes containing about N/50 AgNO3.A current of 7 amps was required to give a sufficient rapid evolution of H2. The current was run for one minute; after that it was switched off and the arsenic-containing solution was introduced through the funnel. The current was then switched on for 15 minutes; darkening of the AgNO3 occurred in half a minute when 1 mg of As was in the apparatus. Usually the second AgNO3 tube was unaffected, however fast the current of gas had been, and its contents were not included in the titration unless a visible amount of metallic Ag had been produced, since even 0.01 mg of AsH3 produced a marked coloration. For the final titration the contents of the first AgNO3 tube were slightlydiluted, given a pinch of bicarbonate, treated with KI in large excess (to dissolve the AgI which is soluble in concentrated KI) and titrated with very dilute I2, using starch which at the endpoint, instead of blue, gave a violet-brown colour in the presence of KI.
8. Cyanide Analysis
In the early 1900s, the test for HCN was the guaiacum-copper test:13 if a tincture of guaiacum (a greenish resin from a South American tree used in medicine) was added to a very dilute aqueous solution of HCN, a white precipitate was formed; if now a few drops of CuSO4 solution was added, a blue colour was produced, rendered more intense by the addition of rectified spirits. However, this test was not characteristic, since other agents also produced a blue colour with it. James Moir did some research on this and suggested that this particular test for HCN could be improved by substituting for the guaiacum certain derivatives of diphenyl compounds which give intense colours upon oxidation. Later he found that 3-3' dimethylbenzidine was by far the best reagent.14 A solution was prepared containing this compound together with copper acetate and acetic acid. When a filter paper moistened with a few drops of this solution was suspended in the air suspected of containing HCN a Prussian-blue colour would appear at once if the HCN present was present in a concentration more than 1 part in 300 000 of air; even with 1 part in 2 000 000 the colour appeared faintly after a minute.
Liquids suspected of containing cyanide were placed in a suitable corked bottle, acidified with H2SO4 and warmed to about 40 °C; the test was then applied to the air in the bottle. He described the reaction as follows:
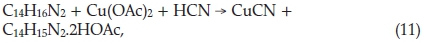
and he claimed that the blue colour would be due to the acetate of the quinhydrone of 3-3'dimethylbenzidine.
9. Chromium Ore Analysis in the Presence of Iron
James Moir made a detailed study of all the errors possible when analyzing chromium ores containing iron.15 He maintained that "...almost every step in the standard methods was fraught with pitfalls, and that the final result could be in error by as much as 10 % of the reported value, depending on the method employed and the analyst who gets results near the truth is only saved by a compensation of errors." He made a series of experiments in which known amounts of chromium were submitted to all the separate stages of the various standard methods of the analysis. He summarized his findings as follows:
a) Errors in the final titration and the use of pipettes and burettes: titrating chromium in the presence of excess standard iron against KMnO4 gives an endpoint which is not clear due to the green colour of the chromium. He suggested various ways of improving the endpoint such as viewing the titration through a glass container filled with acid water faintly coloured with methyl orange: the pink colour changes the green tint to yellow, which at the endpoint changes to the red colour of permanganate. He also stated that it was not possible in any titration process using 20-30 cm3 of reagents to get results any nearer than 0.30 %.
b) Effect of undestroyed sodium peroxide: he showed that when adding sodium peroxide to an alkaline solution of chromium, 10 minutes boiling was insufficient to destroy all the peroxide and he suggested longer boiling, say 20 minutes.
c) Problems caused by the presence of Fe(OH)3: he showed that some chromium could be trapped within the Fe(OH)3 precipitate due to adsorption, partly due to the formation of insoluble ferric chromate, and partly due to incomplete oxidation of Cr(OH)3 upon further boiling with peroxide and subsequent filtering off the Fe(OH)3. He suggested that the ferric hydrate decomposed Na2O2 catalytically at a temperature similar to that at which Na2O2 oxidizes Cr(OH)3, consequently a fraction of the latter remained in the precipitate. Thus in an ordinary peroxide fusion, if the Cr had not been fully oxidized by the fusion, it was not possible to remedy this error by adding peroxide to the aqueous solution, because Fe(OH)3 was always present.
d) Errors in fusions: doing a peroxide fusion in a Ni crucible caused the formation of much Ni2O3, which causes an error when the melt is worked up. Carrying out the fusion in an iron crucible indicated that iron does not reduce chromium in the melt as nickel seems to and it does not form a peroxide and therefore could not interfere later on. With reference to nitre-fusions, he recommended KNO2 over KNO3 in the fusion mixture as there was less effervescence. At the end, whichever nitre was used, a quantity of nitrite remained with the chromate in the melt and when the solution was acidified, the nitrous acid partly destroyed the chromic acid, CrO3 becoming Cr(NO3)3. This problem was overcome by heating the acid solution with excess persulphate until all HNO2 had become HNO3 and all Cr was oxidized, and then boiling for half an hour to destroy excess persulphate; the nitre fusion then gave the correct result. He further analyzed errors in the mercurous chromate method, where HgNO3 was used to precipitate chromates in the presence of high quantities of HNO3, an excess of reagent being required, since it often contained Hg(NO3)2. He pointed out several precautions when using this method.
In order to overcome the problems caused by precipitates, filtrations and reducing agents, he developed a new peroxide-persulphate method for chromite analysis.15 The sample was fused with Na2O2, diluted with water and acidified with 20 % H2SO4 to dissolve the precipitate: potassium persulphate was then added and the solution warmed to 80 °C. Everything should be in solution now, except silica. The chromium, which was partly reduced by acidifying in the presence of H2O2, now started oxidizing to chromic acid. The excess persulphate was then destroyed by adding more 20 % H2SO4 and the mixture was boiled for half an hour (persulphate is stable at 90 °C if not very acid); the solution was now cooled and titrated as usual.
10. A Sensitive Test for Phenol
James Moir developed a sensitive test for phenol.16 On the basis of previous work and having detected phenol in soap when its concentration was less than 0.01 %, he developed a sensitive method which did not require distilling the sample. The reagent used was p-nitroaniline hydrochloride. For a test, the solution, suspected of containing phenol, was first neutralized and some sodium acetate was added. In the meantime 5 cm3 of the yellow p-nitroaniline reagent was treated with dilute sodium nitrite until it became colourless. This mixture was then added to the test fluid, resulting in the development of a pale yellow flesh-colour; an orange precipitate was obtained if much phenol was present. After a minute, excess NaOH was added and a deep colour was obtained: ruby-red with much phenol, rose-red if the phenol was over 0.01 %, and salmon-coloured if it was only present in a few ppm. The latter colour gave a broad absorption band at λ 494 in the spectroscope, where phenol could be distinguished from cresols and other substances which also give a colour with the reagent. The phenol colour is due to a compound he called 'nitrobenzeneazophenol' which in the presence of NaOH is ionized, giving a deep colour on account of its peculiar constitution. Using this test it was possible to detect phenol in a diluted solution of 1 ppm.
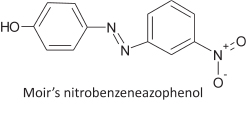
11. Chemical Detective Work
In 1921 James Moir was involved in a case submitted by the Medical Officer for Health: it involved the sneezing-powder 'Catchoo': even a tiny trace, when sniffed into the nostrils, produced uncontrollable sneezing.17 He was asked to identify the material. Preliminary work showed the material to contain a few percent of an active substance, which was the hydrochloride of a white crystalline base melting at 134 °C; the rest was found to be clay. He found initially that the base resembled an alkaloid, but it did not display the reactions of any alkaloid. The main colour reaction was an intense blood-red obtained in acid solution either with FeCl3 or CrO3, evidently due to oxidation. Also, when treated with HAuCl4 a deep blue colour was obtained. From previous experience, he suspected it to be a benzene-type diamino-compound. Consulting his reference book, Mulliken's Identification of Pure Organic Compounds he found 15 possible substances, with melting points between 133 and 140 °C. Of these only five gave the colour with FeCl3. p-Phenylenediamine was tried first as being the likeliest, but it was found to be different from the unknown. The next possibility was 3-amino-p-cresol, melting at 135 °C and giving the red colour with FeCl3; again, it proved different from the unknown, since its HCl salt did not burn the tongue (as did the unknown) and its FeCl3 colour was not as intense as that of the unknown. The other three remaining possibilities appeared to be unlikely from their great rarity. He therefore concluded that Mulliken had not catalogued the substance.
In further work he found that, when he treated the unknown with nitrous acid, the diazo-solution was extremely stable, and could be boiled for five minutes without change. From his extensive research in organic chemistry, he remembered that benzidine (structure shown below) also showed this azo-stability. He therefore tried his bichromate test for benzidine on the unknown, which, although not benzidine, gave a similar indigo-blue precipitate. He thus determined that it was a substitution product of benzidine and, going back to the HAuCl4 coloration, found a similar blue colour; FeCl3 also gave an intense blood-red colour when dissolved in HCl. He found that the substance was dimethoxybenzidine (structure shown below), known in industry as dianisidine, a substance manufactured by the ton for making dyes. It gave a melting point of 135 °C and the azo-dye made from it with phenol had the same spectrum as the one made from 'Catchoo'. Wondering why he hadn't found it in Mulliken, he consulted the reference again and found dianisidine out of place with the wrong melting point!
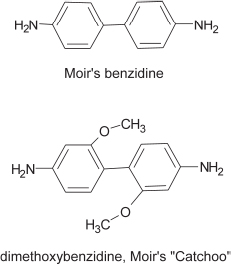
12. Conclusion
This article shows that James Moir was indeed a widely experienced and capable chemical analyst, although most of his research dealt with pure organic chemistry. He was at the forefront of the development of chemical analysis, and in the absence of modern instrumentation, had to resort to fundamental reasoning and much 'wet chemistry work', to solve problems at hand. The South African Chemical Institute allocates each year a 'James Moir Medal' to the best Chemistry Honours or BTech student of a University and in this way he can be remembered as an exceptional pioneering chemist who did so much for chemistry in South Africa in the early 1900s.
References
1 SACI Proceedings 1928-1929, September 1929, pp. 29-30. [ Links ]
2 P. Bloom, James Moir Medal Information, SACI, private communication. [ Links ]
3 J. Moir, J. Chem. Soc, Trans. 1924,125, 1134-1141. [ Links ]
4 SACI Proceedings, June 1916, pp. 24-31. [ Links ]
5 J. Moir, J. S.A. Chem. Inst., 1925, 8, 2, 3-7. [ Links ]
6 J. Moir, J. S.A. Assoc. Anal. Chem., 1921, 4, 1, 3-8. [ Links ]
7 J. Moir, J. S.A. Chem. Inst.,1923, 6, 2, 69-71. [ Links ]
8 J. Moir, J. S.A. Assoc. Anal. Chem., 1920, 3, 2, 6-8. [ Links ]
9 J. Moir, J. S.A. Assoc. Anal. Chem., 1918,1, 1, 18-20. [ Links ]
10 J. Moir, J. S.A. Chem. Inst., 1928,11, 1, 33. [ Links ]
11 H.H. Green, J. S.A. Assoc. Anal. Chem., 1920, 3, 2, 3-4. [ Links ]
12 J. Moir, J. S.A. Chem. Inst., 1925, 8, 2, 16-18. [ Links ]
13 J. Pereira, The Elements of Materia Medica, Longmans, London, 1839, p. 241. [ Links ]
14 J. Moir, J. S.A. Assoc. Anal. Chem., 1920, 3, 2, 16. [ Links ]
15 J. Moir, J. S.A. Assoc. Anal. Chem., 1919, 2, 1, 9-16. [ Links ]
16 J. Moir, J. S.A. Chem. Inst., 1922, 5, 1, 8-9. [ Links ]
17 J. Moir, J. S.A. Assoc. Anal. Chem., 1921, 4, 2, 15-19. [ Links ]
Received 17 October 2013
Accepted 26 March 2014
* E-mail: ployson@nmmu.ac.za


{kind=link}