










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban ene. 2014
RESEARCH ARTICLE
Rhenium(V) complexes containing mono- and tridentate imido ligands: crystal structures, spectroscopic results and DFT optimization
B. SchmittI; N. SabanciII; B. DedeogluIII; V. AviyenteIII, *; E. HostenI; T.I.A. GerberI, *; G. Habarurema
IDepartment of Chemistry, Nelson Mandela Metropolitan University, Port Elizabeth, South Africa
IIDepartment of Chemistry, Siirt University, 56100, Siirt, Turkey
IIIDepartment of Chemistry, Bogazici University, 34342, Istanbul, Turkey
ABSTRACT
Rhenium(V) complexes containing the [ReX(PPh3)2]4+ (X = Br, I) moiety were studied. The reaction of N-(2-aminophenyl)sali-cylideneimine (H3pna) with irans-[ReOBr3(PPh3)2] produced the complex salt [Re(pna)Br(PPh3)2]0.5Br.0.5(ReO4) (1), in which the tridentate ligand pna is coordinated via a doubly deprotonated nitrogen (as an imide), an imino nitrogen and a deprotonated phenolate oxygen atom. The reaction of frans-[ReO(OEt)I2(PPh3)2] with two equivalents of 2-aminophenol (H3ap) in ethanol led to the isolation of the '2+1' complex salt [Re(Hap)(H2ap)I(PPh3)2]I (2) in good yield. The Hap ligand is coordinated monodentately via the doubly deprotonated imido nitrogen, and H2ap is chelated bidentately through the neutral amino nitrogen and a deprotonated phenolate oxygen atom. The crystal structures of 1 and 2 were determined by X-ray single crystal diffraction. Spectroscopic results and DFT calculations are also reported.
Keywords: Rhenium(V), tridentate imido, '2 + 1' coordination, X-ray crystal structures, DFT.
1. Introduction
The coordination chemistry of the metal rhenium is dominated by the +V oxidation state, since it can easily be accessed from the two-electron reduction of perrhenate (ReO4-). Such oxorhenium(V) complexes are usually stable and substitutionally inert, making them ideally suited as building blocks for potential therapeutic applications in nuclear medicine.1 One of the disadvantages of this approach, however, is that the oxo group dominates the chemistry of rhenium(V) in terms of structures, geometries, reactivity and magnetic properties, which limit the further exploration of the coordination chemistry in this oxidation state. For this reason many current research efforts focus on rhenium(V) cores with metal-nitrogen multiple bonds, such as metal(V)-nitrido, hydrazido and imido moieties.2-5
Part of our current focus is on stable Re(V) complexes with multidentate ligands containing an amino group, which on deprotonation can lead to coordinated chelates containing the phenylimido [Re=NC6H4-X] core, or other coordinated derivatives of nitrogen, such as imines, amines and amides. One of the advantages of the phenylimido group over the oxo group is that it can be functionalized and derivatized. For example, the compound [188Re(=NC6H4-X)Cl3(PPh3)2] contains a para-substituted phenylimido group which was functionalized with a carboxylic group conjugated to various cholesterol derivatives.6 However, it has also been shown that many phenylimidorhenium(V) compounds are hydrolytically unstable, which was ascribed to the presence of monodentate ligands in the coordination sphere.7 For this reason, the use of multidentate imido-containing ligands in the synthesis of rhenium(V) complexes may be advantageous. We have recently reported the synthesis and structure of the rhenium(V) complex [Re(amb)(OEt)I(PPh3)2], in which the trianionic bidentate 2-imido-3-methylbenzoate (amb3-) chelates via the imido nitrogen and deprotonated acetoxy oxygen.8
We have now extended this to the synthesis of a rhenium(V) complex containing a tridentate ligand with a coordinated imido nitrogen. In this account, two rhenium(V) complex salts of the core [ReX(PPh3)2]4+ (X = Br, I), containing a coordinated imido nitrogen, are reported. In the complex cation [Re(pna)Br(PPh3)2]+ the trinegative tridentate ligand pna3- (H3pna = N-(2-amino-phenyl)salicylideneimine) chelates through the dinegative imido nitrogen, the imino nitrogen and the phenolate oxygen atoms. In [Re(Hap)(H2ap)I(PPh3)2]+ (H3ap = 2-aminophenol) the three coordination sites occupied by pna3- have been replaced by Hap2-, which is coordinated in a monodentate fashion via the doubly deprotonated imido nitrogen, and H2ap which is coordinated bidentately via a singly deprotonated amido nitrogen and phenolate oxygen.
2. Experimental
2.1. General Procedure
Trans-[ReOBr3(PPh3)2] and trans-[ReO(OEt)I2(PPh3)2] were prepared according to literature procedures.9,10 Scientific instrumentation used is the same as reported elsewhere.11 Infrared spectra were obtained using KBr discs and 1H NMR spectra were recorded in d6-DMSO. 2-Aminophenol (H3ap) and all other chemicals were obtained commercially (Aldrich). The ligand N-(2-aminophenyl)salicylideneimine (H3pna) was synthesized utilizing the Schiff base condensation reaction of salicylaldehyde with an equimolar amount of 1,2-diaminobenzine in ethanol. The IR and NMR spectra of the two novel complexes are provided with the online supplement.
2.2. Synthesis of [Re(pna)Br(PPh3)2]0.50Br.0.50ReO4 (1)
A mixture of trans-[ReOBr3(PPh3)2] (110 mg, 117 μmol) and H3pna (23 mg, 117 μmol) was heated under reflux in 20 cm3 of ethanol for 2h. After cooling to room temperature, the volume was reduced to ~5 cm3 under vacuum, and a green precipitate was removed from the solution. The filtrate was left to evaporate slowly at room temperature, and after two days green crystals were removed by filtration. Yield = 52 % (based on Re), m.p. 189 °C. Anal. Calcd. (%) for 2(C49H39BrN2OP2Re).ReO4.Br: C, 50.5; H, 3.4; N, 2.4. Found: C,50.7; H, 3.6; N,2.3. IR (nmax/cm-1): n(Re=N) 1092s; n(c=N) 1587s, 1610s; n(ReVII=O) 905s; n(Re-N)518m; n(Re-O) 443m. 1H NMR (295K, ppm, CDCl3): 13.52 (s, 1H, H(1)), 7.61-7.30 (m, 31H, PPh3, H(123)), 7.18 (d, 1H, H(113)), 7.06 (t, 1H, H(124)), 6.91 (t, 1H, H(114)), 6.80 (d, 1H, H(126)), 6.73 (d, 1H, h(116)), 6.62 (t, 1H, H(125)), 6.48 (t, 1H, H(115)). UV-Vis (DMF, lmax (e,M-1 cm-1)): 293(7300), 407 (980),471(230). Conductivity (10-3M, CH3CN) = 42 ohm-1 cm2 mol-1.
2.3. Synthesis of [Re(Hap)(H2ap)I(PPh3)2]I (2)
H3ap (0.50 g, 260 μmol) was added to a solution of trans-[ReO(OEt)I2(PPh3)2] (133 mg, 130 μmol) in 15 cm3 ethanol. The mixture was heated under reflux for 3 h. After cooling to room temperature, the solution was filtered, and the filtrate was left to evaporate slowly over a period of two days, after which green crystals, suitable for X-ray diffraction studies, were collected. Yield = 57 %, m.p. 232 °C. Anal. Calcd. (%) for C48H40I2N2O2P2Re: C,48.9; H, 3.4; N, 2.4. Found: C, 48.7; H, 3.7; N, 2.6. IR (cm-1): n(Re=N) 1091s; n(N-H) 3224w, 3251w; n(Re-N) 456m; n(Re-O) 448m. 1H NMR (295K, ppm): 7.21 (m, 16H, PPh3); 7.33 (m, 8H, PPh3); 7.49 (m, 6H, PPh3); 7.61 (m, 2H, H(113), H(116)); 6.87 (d, 1H, H(12ó)); 6.73 (t, 2H, H(114), H(115)); 6.68 (d, 1H, H(123)); 6.31 (t, 2H, H(124), H(125)). Electronic spectrum (CHCl3,λ (ε,M-1 cm-1)): 433 (2260), 394 (2500), 301 (6200). Conductivity (10-3 M, CH3CN) = 37 ohm-1 cm2 mol-1.
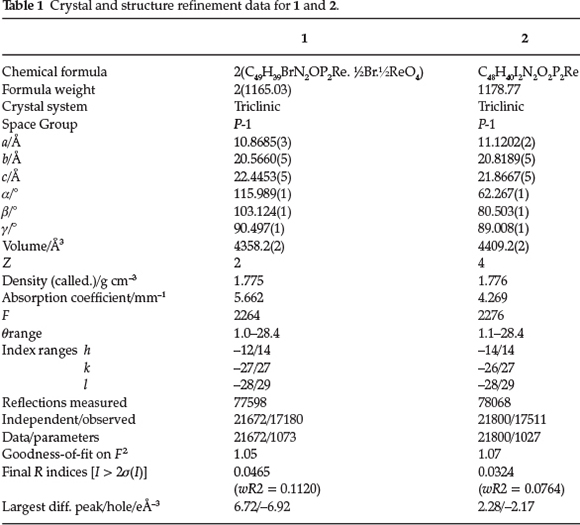
2.4. X-ray Crystallography
X-ray diffraction studies of 1 and 2 were performed at 200 K using a Bruker Kappa Apex II diffractometer with graphite monochromated Mo Ka radiation (l = 0.71073 A). APEXiI was used for data collection and SAINT for cell refinement and data reduction.12 The structures were solved by direct methods using SHELXS-2013,13 and refined by least-squares procedures using SHELXL-201313 with SHELXLE14 as a graphical interface. All non-hydrogen atoms were refined anisotropically, and the hydrogen atoms were calculated in idealized geometrical positions. Data were corrected for absorption effects by the numerical methods using SADABS.12 Reflections obscured by the beam stop were omitted from the refinement. For structure 1 the position of the bromide counter-ion was found to be positionally delocalized with a 0.782:0.218 ratio. The crystal and structure refinement data for the compounds are given in Table 1 (see Figs 1 and 2 for the molecular structures). The X-ray data were deposited with the Cambridge Crystallographic Data Centre and the details are provided in the online sSupplement.
2.5. Computational Details
The gas phase geometries of complexes 1 and 2 were optimized with DFT using the B3LYP15-17 and PBE1PBE18 functionals of Gaussian 09.19 For both functionals, the calculations were performed by using the ECP basis set (LANL2DZ)20 for rhenium, 6-31G21-30 for bromine, carbon, oxygen, nitrogen and hydrogen, 3-21G for iodine31-36 and 6-31+G(d)37-38 for phosphorus. An additional d function with exponent a = 0.3811 and an f function with exponent a = 2.0330 on rhenium were added.21,38 The positive vibrational frequencies of the optimized structures have confirmed the optimized structures as being true minima. The electronic energy gaps between the filled and empty orbitals have been obtained with the time-dependent density functional btheory (TD-DFT)39-45 in a polar medium described by a dielectric constant (e = 36.7 for DMF and e = 4.81 for CHCl3) (Figs. 3-4). The PCM methodology46-17 was used to take into account the effect of the medium. Chemcraft was used to reproduce the molecular orbitals (http://www.chemcraftprog.com/) in Figs. 5, 6, S1 and S2. The Cartesian coordinates of the optimized structures are provided as supplementary material.
3. Results and Discussion
3.1. [Re(pna)Br(PPh3)2]0.50Br.0.50ReO4 (1)
The ligand H3pna was synthesized by the condensation of equimolar quantities of salicylaldehyde and 1,2-diamino-benzene in ethanol. 1 was prepared by the reaction of fraws-[ReOBr3(PPh3)2] with two mol equivalents of H3pna in ethanol,
[ReOBr3(PPh3)2] + H3pna → [Re(pna)Br(PPh3)2]+ + HBr + Br- + H2O.
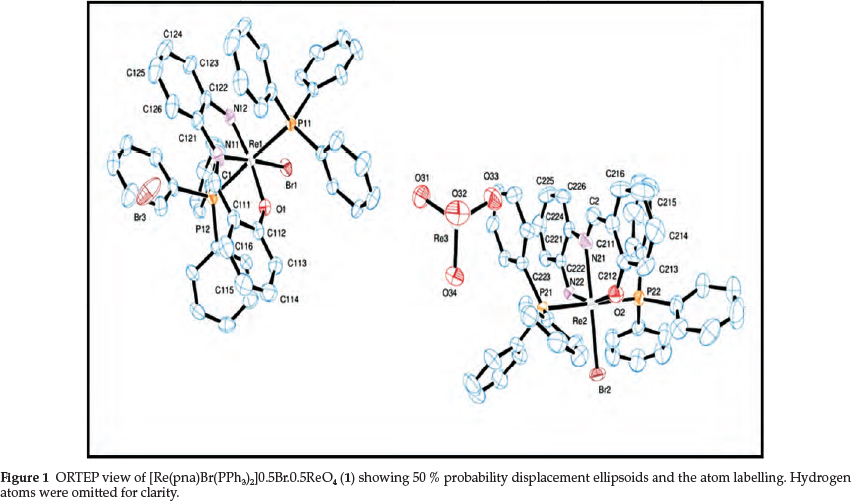
X-ray crystallographic and spectroscopic results show that two independent and identical novel rhenium(V)-imido complex salts with bromide and perrhenate counter-ions were isolated. The chelate pna3- is present in 1 as a trinegative tridentate ligand, with coordination through the doubly deprotonated amino nitrogen (to form a coordinated imido group), the imine nitrogen and the deprotonated phenolic oxygen atoms. Due to the propensity of rhenium(V) to form neutral complexes, the formation of the cation [Re(pna)Br(PPh3)2]+ is quite surprising, since neutrality could easily have been obtained by substitution of PPh3 ligand to form [Re(pna)Br2(PPh3)]. The formation of the [ReO4]~ counter-ion is also surprising, and it intimates that complex redox processes are operative in this reaction system.
Complex 1 is diamagnetic and a 1:1 electrolyte in acetonitrile, and it is only soluble in the polar solvents DMF, DMSO and acetonitrile. The IR spectrum of 1 displays the Re=N stretching frequency as a medium-intensity band at 1092 cm-1, with no band in the 920-990 cm-1 region that can be ascribed to n(ReV = O). The presence of the perrhenate is indicated by the very strong peak centred at 905 cm-1 [n(ReVI1 = O)]. Re-N and Re-O stretches occur at 518 and 443 cm-1, respectively. Although the 1H NMR spectrum of the complex is dominated by signals of the phosphine protons, it is quite informative on the coordination mode of the pna ligand. The methine proton on C1/C2 (see Fig. 1) appears as a singlet the furthest downfield at 13.52 ppm, and all the phenyl protons of pna could be identified by clear signals (see section 2.2 and Fig. 1). The electronic spectrum in DMF illustrates three absorptions at 293, 407 and 471 nm. With reference to previous spectroscopic studies,48 the band at 293 nm is tentatively assigned to intraligand π → π* and LMCT (ligand-to-metal charge transfer) transitions. The two other absorptions are ascribed to a LMCT and a d-d transition, respectively.
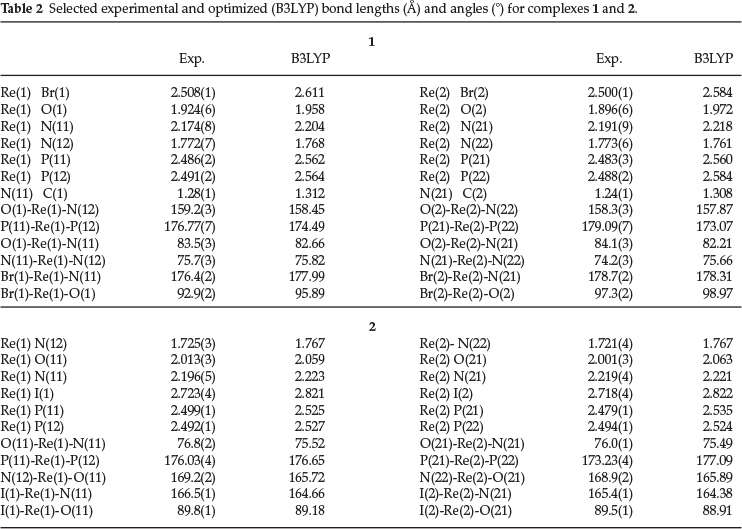
Fig. 1 shows that there are two molecules of [Re(pna)Br(PPh3)2]+ in the asymmetric unit, together with the two counter-ions Brand [ReO4]-. The bonding parameters of the two molecules are very similar. Each rhenium(V) ion is centred in an octahedral environment with the equatorial plane formed by the P2BrN donor set. The octahedron is severely distorted, with the Br-Re-P bond angles close to orthogonality [Br(1)-Re(1)-P(11) = 91.01(5) °, Br(1)-Re(1)-P(12) = 90.99(5) °, Br(2)-Re(2)-P(21) = 90.56(6) °, Br(2)-Re(2)-P(22) = 89.47(6) °]. There are large deviations from orthogonality for the Br-Re-O [Br(1)-Re(1)-O(1) = 92.9(2) °, Br(2)-Re(2)-O(2) = 97.3(2) °], Br(1)-Re(1)-N(12) [107.9(2) °] and Br(2)-Re(2)-N(22) [104.4(2) °] angles. The N(12)-Re(1)-O(1) [159.2(3) °] and N(22)-Re(2)-O(2) [158.3(3) °] angles deviate considerably from linearity. The P-Re-P angles are 176.77(7) ° and 179.09(7) ° for 1 and 2, respectively. The bite angles O(1)-Re(1)-N(11) [83.5(3) °] and N(11)-Re(1)-N(12) [75.7(3) °] are comparable to those in 2 [84.1(3) ° and 74.2(3) °, respectively].
The pna ligand acts as a terdentate trianionic moiety, with N(12) and N(22) coordinated to the metal as dinegative imido nitrogen atoms. The Re(1)-N(12) and Re(2)-N(22) bond lengths are identical [1.772(7) A], and fall at the upper limit of the range normally observed for Re-N(imido) bonds.2,49 They are considerably shorter than the values usually found for ReV-amide [1.98-2.05 A] and ReV-aminobonds [2.15-2.23 A].50 The Re(1)-N(12)-C(122) [129.3(6) °] and Re(2)-N(22)-C(222) angles [131.0(5) °] indicate significant deviation from linearity of the coordination mode of the phenylimido unit, with a concomitant reduction in bond order. The Re(1)-N(11) [2.174(8) A] and Re(2)-N(21) [2.191(9) A] bond lengths are typical of ReV-N(imine) bonds,50 and the Re-O lengths [Re(1)-O(1) = 1.924(6) A, Re(2)-O(2) = 1.896(6) A] fall at the lower end of the range normally observed for ReV-O (phenolate) bonds.51
The average C-N-C bond angle around N(11) and N(21) is 119.7(9) °, close to the ideal of 120 ° for a sp2 hybridized nitrogen atom. The N(11)-C(1) [1.28(1) A] and N(21)-C(2) bonds [1.24(1) A] are double.
The [ReO4]- counter-ion has approximately regular tetrahedral geometry, with the average Re(3)-O distance equalling 1.715(8) A. Both the two counter-ions make close contacts with both complex cations.
It was shown previously that the reaction of 1,2-diamino-benzene (H2dab) with fraws-[ReOCl3(PPh3)2] in ethanol produces fraws-[Re(dab)Cl3(PPh3)2], in which dab is coordinated as a monodentate imide.52 The study was extended to multi-dentate ligands containing an amino group which, on deprotonation, can lead to a coordinated chelate containing the phenylimido core. The structure of the rhenium(V) complex [Re(amb)(OEt)I(PPh3)2], in which the trianionic bidentate 2-imido-3-methylbenzoate (amb3-) chelates via the imido nitrogen and acetoxy oxygen, was recently reported.8 From the reduction of [ReO4]- with triphenylphosphine in the presence of H3pna and hydrochloric acid, the neutral complex [ReV(pna)Cl2(PPh3)] was isolated.53
3.2. [Re(Hap)(H2ap)I(PPh3)2]I (2)
The oxo-free complex cation fraws-[ReV(Hap)(H2ap)I(PPh3)2]+ was isolated as the iodide salt (2) from a 1:2 molar ratio reaction between fraws-[ReO(OEt)I2(PPh3)2] and aminophenol (H3ap) in ethanol, according to the equation
[ReO(OEt)I2(PPh3)2] + 2H3ap ® 2 + H2O + EtOH
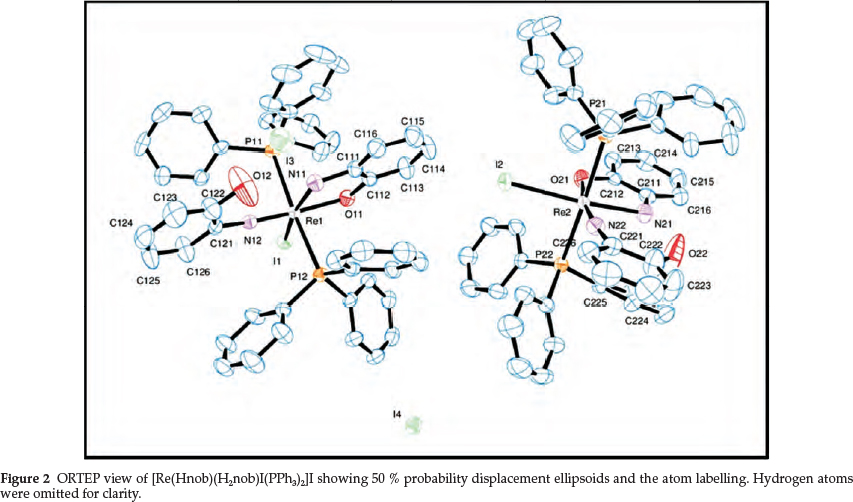
Complex 2 is soluble in most polar solvents like DMSO, DMF, acetonitrile and chloroform. It is a 1:1 electrolyte in DMF. The IR spectrum contains a medium-intensity absorption at 1091 cm-1, which is within the region (1000-1200 cm-1) expected for n(Re = N).2,49 Two weak peaks at 3224 and 3251 cm-1 are indicative of the presence of the neutral amino group in the complex. The 1H NMR spectrum is dominated by three multiplets (at 7.21, 7.33 and 7.49 ppm) due to the 30 protons of the two PPh3 groups. These three peaks integrate for 16, 8 and 6 protons, respectively. The eight aromatic protons of the Hap and H2ap ligands give rise to a two-proton multiplet (at 7.61 ppm), a one-proton doublet at 6.87 ppm, a triplet at 6.73 ppm (two protons), a doublet at 6.68 ppm (one proton) and a two-proton triplet at 6.31 ppm. These signals have been tentatively assigned to the protons H(113)/H(116), H(126), H(114)/H(115), H(123), and H(124)/H(125), respectively (see Fig. 2 for labelling). The UV-vis spectrum in chloroform exhibits two electronic transitions at 394 and 301 nm due to intraligand π ® π* and LMCT transitions. A d-d transition is indicated by a weaker absorption at 433 nm.
There are two molecules of the complex cation [Re(Hap) (H2ap)I(PPh3)2]+ in the asymmetric unit (Fig. 2). Each Re(V) ion resides in a distorted octahedral environment, with the largest contributors to the distortion being the angles N(12)-Re(1)-I(1) [101.0(1) °] and N(12)-Re(1)-O(11) [169.2(2) °] in the Re(1) complex [N(22)-Re(2)-I(2) = 101.6(2) °; N(22)-Re(2)-O(21) = 168.9(2) °]. The N(12)-Re(1)-P(11) [92.3(1) °], N(12)-Re(1)-P(12) [91.6(1) °] and N(12)-Re(1)-N(11) [92.5(2) °] angles are close to orthogonality, and the P(11)-Re(1)-P(12) angle is 176.03(4) °, significantly larger than the P(21)-Re(2)-P(22) one [173.23(4) °].
The Hap ligand is coordinated monodentately to the metal via the imido nitrogens N(12) and N(22) in the two molecules, respectively, with the respective Re(1)-N(12) [1.725(3) A] and Re(2)-N(22) [1.721(4) A] bond distances typical of rhenium(V)-imido bonds.249 The Re(1)-N(12)-C(121) [169.0(4) °] and Re(2)-N(22)-C(221) [171.0(4)°] angles are substantially larger than the corresponding angles in complex 1, resulting in increased bond order and considerably shorter Re-N(imido) bond distances in 2.
The H2ap ligand acts as a bidentate monoanionic chelate. The Re(1)-O2(11) [2.013(3) A] and Re(2)-O(21) [2.001(3) A] bond distances are longer than these bonds in complex 1, with the Re(1)-O(11)-C(112) [120.1(3) °] and Re(2)-O(21)-C(212) [120.7(3) °] angles substantially smaller than in 1 [Re(1)-O(1)-C(112) = 138.6(6)°; Re(2)-O(2)-C(212) = 138.1(6)°]. The Re(1)-N(11) [2.196(5) A] and Re(2)-N(21) [2.219(4) A] bond lengths are longer than the Re-N(imino) lengths in 1, and they are typical of Re(V)-N(amino) bonds. The bite angles O(11)-Re(1)-N(11) and O(21)-Re(2)-N(21) are 76.8(2) ° and 76.0(1) °, respectively.
The compound [Re(ddd)(Hddd)I(PPh3)2](ReO4) was isolated from the reaction of cis-[ReO2I(PPh3)2] with the uracil derivative 5,6-diamino-1,3-dimethyl-2,4-dioxopyrimidine (H2ddd).54 The X-ray crystal structure shows that ddd is coordinated mono-dentately as an imide, and Hddd as a bidentate via the amino and amido nitrogen atoms, very similar to 2.
3.3. Geometry Optimizations and Characterization of the Molecular Orbitals
The X-ray data have been considered to draw the initial inputs for 1 and 2 and these were optimized in a singlet state with the B3LYP and PBE1PBE functionals, and the optimized geometric parameters of the complexes are summarized in Tables 2 and S1, respectively. In general, the calculated bond lengths and angles agree well with the experimental data. The Re-Br and Re-P(1) and Re-I(2) bond lengths show the largest deviations. This may be due to the basis sets which are approximated to some extent, or it may be due to the influence of the crystal packing in experiment as opposed to vacuum in the calculations.
With the B3LYP functional, the highest occupied MO's of 1 are of the dxz type (with some contributions from the pπ oxygen and pna3- orbitals and π orbitals of Br. The LUMO and LUMO+1 are predominantly localized on the rhenium atom (dxy and dyz orbitals, respectively) with some contribution of the π orbitals of imido nitrogen, bromide and phenoxide oxygen to the former and of the pp orbitals of imido nitrogen and pna3- orbitals to the latter. The LUMO+3 is of dz2 character, with a contribution of the π orbitals of PPh3 (Fig. 3).
The qualitative MO scheme of 2 is similar to that of 1. The HOMO is of the dxz type with a contribution from the pπ orbital of iodide. The LUMO is entirely localized on the rhenium atom (dxy). The LUMO+1 (dyz) and LUMO+2 (dxz) are predominantly located on the rhenium atom, with some contribution from the imido nitrogen (pπ) to the former and pπ (PPh3) to the latter. The LUMO+4 is of dz2 character (Fig. 4).
With the PBE1PBE functional in 1, the HOMO is centered on the dxz orbital, with π contributions from the ligands pna3-, bromide and the imido nitrogen. The LUMO consists of the dxy and the π-orbitals of pna3-. The LUMO+3 is localized on the dxz orbital of the metal, with contributions from the π and π* orbitals of PPh3. The orbitals for complex 2 are very similar to those predicted with the B3LYP functional. The HOMO is also located on the dxz orbital, with a contribution from iodide's pπ orbital. The LUMO is also entirely located on the dxy of rhenium, and the LUMO+1 is primarily localized on the dyz with some contribution from the π-orbital of the imido nitrogen of pna3-(Figs S1 and S2).
3.4. Electronic Spectra
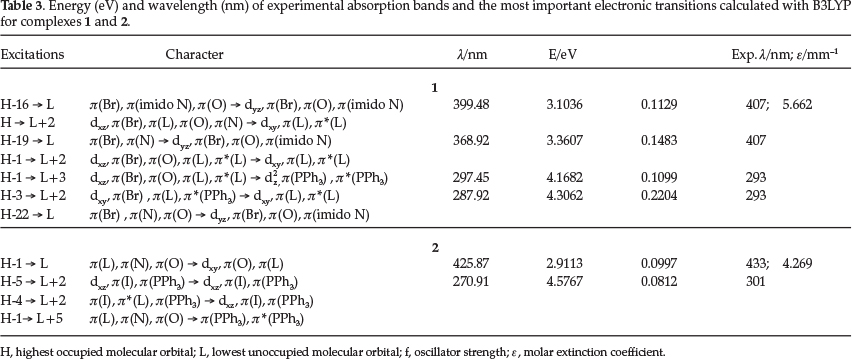
The assignment of the calculated excitations to the experimental bands was based on the contour plots and relative energy of the HOMOs and LUMOs (Table 3, Figs 3-4 and Figs S1-S2) involved in the electronic transitions.
The B3LYP calculations show that the experimental band of 1 at 407 nm may originate from the HOMO to LUMO + 2 and HOMO-16 to LUMO transitions. As can be seen from Fig. 3, the LUMO and LUMO+2 orbitals are mainly formed from the rhenium dxy orbital. The HOMO and HOMO-16 are localized amongst the rhenium ion, phenoxide oxygen and pna-3 ligand. Accordingly, the transitions assigned to this experimental wavelength can be considered as mixed phenoxide/rhenium LMCT and d-d LF (ligand field) transitions.
The experimental absorption band at 293 nm results mainly from LMCT and interligand (IL) transitions, and d-d transitions.
For 2 the highest wavelength experimental band at 433 nm originates from the HOMO-1 and hOmO-2 to LUMO transitions. The LUMO is composed of the metal's dxy orbital, the HOMO-1 is formed from the delocalization amongst the ligand H2nob and imido nitrogen atom, and HOMO-2 from the delocalization amongst the iodide and H2nob ligand. The experimental band at 301 nm results from ligand-ligand charge transfer (LLCT), IL transitions, and LMCT from the imido nitrogen and PPh3to the rhenium dxz and dz2 orbitals.
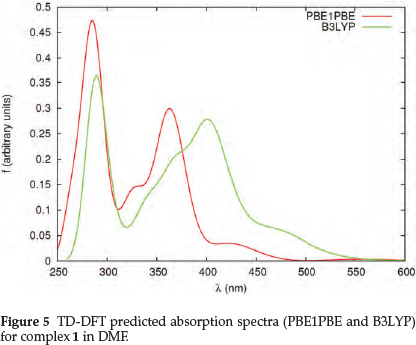
The PBE1PBE calculations show that the energy band of 1 which corresponds to the experimental 407 nm (Fig. 5) is the result of a mixture of HOMO-6 and HOMO-7 to LUMO, and HOMO-16 to LUMO+1 transitions, which originate from IL, d-d, LMCT and LLCT processes. The band which corresponds to the experimental 293 nm results from the HOMO-15 and HOMO-16 to LUMO + 1 transition, and is the result of LMCT and LLCT transitions (Table S2). For 2 the bands at 433 and 301 nm are the result of HOMO-1 to LUMO and HOMO-3 to LUMO+2 transitions (Fig. 6). The former results from LMCT and IL transfers, with the latter originating from LMCT and LLCT processes (Table S2).

3.5. Comparison of the B3LYP and PBE1PBE Functionals against Experimental Data
The root mean square deviations (RMSD) have been calculated for the geometrical parameters (bond lengths and angles) against the experimental values. The RMSD deviations for 1 are 1.5576 with and 1.7531 with B3LYP and PBE1PBE, respectively, whereas 1.4069 and 1.6127 for 2. In both cases the B3LYP functional yields the smallest RMSD. Comparison of the B3LYP and PBE1PBE functionals in terms of the experimental absorption bands reveal the fact that for complex 1 with B3LYP the errors (Δ1 = λ calc - λ t) range between -4 nm < Δλ <8 nm, whereas with PBE1PBE the errors range between -13 nm < Δλ< -36 nm. For complex 2 with B3LYP the errors range between -7 nm < Δλ < -31 nm, whereas with PBE1PBE the errors range between -20 nm < Δλ < -31 nm. Comparison of the B3LYP and PBE1PBE functionals (Figs 3-4) shows that the spectra are shifted to higher wavelengths with B3LYP and yield closer values to the experimental findings. Among the two functionals used in this study, B3LYP seem to reflect a better agreement with experiment.
4. Conclusion
The reaction of N-(2-aminophenyl)salicylideneimine (H3pna) with trans-[ReOBr3(PPh3)2] produced the '3 + 0' complex salt [Re(pna)Br(PPh3)2]0.5Br.0.5(ReO4) (1), in which the tri-dentate O2N-donor ligand pna is coordinated via a doubly deprotonated nitrogen (as an imide), an imino nitrogen and a deprotonated phenolate oxygen atom. The reaction of trans-[ReO(OEt)I2(PPh3)2] with two equivalents of 2-aminophenol (H3ap) in ethanol led to the isolation of the '2 + 1' complex salt [Re(Hap)(H2ap)I (PPh3)2]I (2) in which the Hap ligand is surprisingly coordinated via the doubly deprotonated imido nitrogen only, and H2ap is chelated bidentately through the neutral amino nitrogen and a deprotonated phenolate oxygen atom. The average Re-N(imido) bond length in 1 [average 1.773(6) A] is considerably longer than in 2 [average 1.723(4) A].
Optimization of the X-ray structures of 1 and 2 in a singlet state with the B3LYP and PBE1PBE functionals were done. In general, the calculated bond parameters agree well with the experimental data. In both 1 and 2 the HOMO is located on the dxz orbital, while the LUMO is primarily localized on the dxy orbital of rhenium. Among the two functionals used, B3LYP seems to reflect a better agreement with experimental data.
Acknowledgements
This work was supported financially by the 'Nuclear Technologies in Medicine and the Biosciences Initiative' (NTeMBI), a national technology platform developed and managed by the South African Nuclear Energy Corporation and supported by the Department of Science and Technology. N.S. acknowledges the grant from the TÚBÍTAK BÍDEB 2218-National Postdoctoral Research Scholarship Programme.
Supplementary Material
CCDC 973316 (1) and 973324 (2) contain the supplementary crystallographic data for the complexes. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/re-trieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 IEZ, U.K.; fax: (+044)1223-336-033; or e-mail: depositccdc.cam.ac.uk. The Cartesian coordinates of the optimized structures, the IR and NMR spectra of the two complexes are provided as supplementary material.
The selected experimental and optimized bond lengths (A) and angles (°) for 1 and 2 (PBE1PBE) are given in Table S1 in the online supplement; the energy and wavelengths of experimental absorption bands and the most important electronic transitions (PBE1PBE) for complexes 1 and 2 are given in Table S2. The energy (eV), character and some contours of the molecular orbitals of Complexes 1 and 2 (PBE1PBE) are given in Fig. S1 and Fig. S2, respectively.
References
1 S. Liu and D.S. Edwards, Chem. Rev., 1999, 99, 2235-2268. [ Links ]
2 X. Couillens, M. Gressier, R. Turpin, M. Dartiguenave, Y. Coulais and A.L. Beauchamp,/. Chem. Soc. Dalton Trans., 2002, 914-924. [ Links ]
3 F. Refosco, F. Tisato, C. Bolzati and G. Bandoli, /. Chem. Soc. Dalton Trans., 1993, 605-618, and references therein. [ Links ]
4 U. Abram, Inorg. Chem. Commun., 1999, 2, 227-229. [ Links ]
5 B. Machura, I. Gryca and M. Wolff, Polyhedron, 2012, 31, 128-135. [ Links ]
6 J.B. Arterburn, K.A. Hall, I.M. Fogarty, D.M. Goreham, J.C. Bryan and K.C. Ott, Radiochim. Acta, 1997, 79, 119-122. [ Links ]
7 M. Porchia, F Fisato, F Refosco, C. Bolzati, M. Cavazza-Ceccato and G. Bandoli, Inorg. Chem., 2005, 44, 4766-4776. [ Links ]
8 T.I.A. Gerber, D. Luzipo and P. Mayer, /. Coord. Chem., 2003, 56, 1549-1554. [ Links ]
9 N.P. Johnson, C.J.L. Lock and G. Wilkinson, Inorg. Synth., 1967, 9, 145-148. [ Links ]
10 G.F Ciani, G. Dalfonso, P. Romiti, A. Sironi and M. Freni, Inorg. Chim. Acta, 1983, 72, 29-37. [ Links ]
11 P. Mayer, N. Yumata, T.I.A. Gerber and A. Abrahams, S. Afr. /. Chem., 2010, 63, 40-44. [ Links ]
12 APEX2, SADABS and SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2010. [ Links ]
13 G.M. Sheldrick, Acta Cryst., 2008, A64, 112-122. [ Links ]
14 C.B. Hübschle, G.M. Sheldrick and B. Dittrich, /.Appl. Cryst., 2011,44, 1281-1284. [ Links ]
15 C. Lee, W. Yang and R.G. Parr, Phys. Rev., 1988, B37, 785-789. [ Links ]
16 A.D. Becke, /. Chem. Phys., 1993, 98, 1372-1377. [ Links ]
17 A.D. Becke, /. Chem. Phys., 1993, 98, 5648-5652. [ Links ]
18 C. Adamo and V Barone, /. Chem. Phys., 1999,110, 6158-6170. [ Links ]
19 M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, et al., Gaussian 09. Wallingford CT, Gaussian, Inc., 2009. [ Links ]
20 T.H. Dunning Jr. and P.J. Hay, in Modern Theoretical Chemistry, (H.F. Schaefer III, ed.), vol. 3, Plenum Press, New York, USA, 1977, pp.1-28. [ Links ]
21 R. Ditchfield, W.J. Hehre and J.A. Pople, /. Chem. Phys., 1971, 54, 724-728. [ Links ]
22 W.J. Hehre, R. Ditchfield and J.A. Pople, /. Chem. Phys., 1972, 56, 2257-2261. [ Links ]
23 P.C. Hariharan and J.A. Pople, Theor. Chem. Acc., 1973, 28, 213-222. [ Links ]
24 P.C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209-214. [ Links ]
25 M.S. Gordon, Chem. Phys. Lett., 1980, 76, 163-168. [ Links ]
26 M.M. Francl, W.J. Pietro, W.J. Hehre, J.S. Binkley, D.J. DeFrees, J.A. Pople and M.S. Gordon, /. Chem. Phys., 1982, 77, 3654-3665. [ Links ]
27 R.C. Binning Jr. andL.A. Curtiss, /. Comp. Chem., 1990,11,1206-1216. [ Links ]
28 J.P. Blaudeau, M.P. McGrath, L.A. Curtiss and L. Radom, /. Chem. Phys., 1997,107, 5016-5021. [ Links ]
29 V.A. Rassolov, J.A. Pople, M.A. Ratner and T.L. Windus, /. Chem. Phys., 1998,109, 1223-1229. [ Links ]
30 V.A. Rassolov, M.A. Ratner, J.A. Pople, P.C. Redfern and L.A. Curtiss, /. Comp. Chem., 2001, 22, 976-984. [ Links ]
31 J.S. Binkley, J.A. Pople and W.J. Hehre, /. Am. Chem. Soc., 1980, 102, 939-947. [ Links ]
32 M.S. Gordon, J.S. Binkley, J.A. Pople, W.J. Pietro and W.J. Hehre, /.Am. Chem. Soc., 1982,104, 2797-2803. [ Links ]
33 W.J. Pietro, M.M. Francl, W.J. Hehre, D.J. Defrees, J.A. Pople and J.S. Binkley, /. Am. Chem. Soc., 1982,104, 5039-5048. [ Links ]
34 K.D. Dobbs and W.J. Hehre, /. Comp. Chem., 1986, 7, 359-378. [ Links ]
35 K.D. Dobbs and W.J. Hehre, /. Comp. Chem., 1987, 8, 861-879. [ Links ]
36 K.D. Dobbs and W.J. Hehre, /. Comp. Chem., 1987, 8, 880-893. [ Links ]
37 T. Clark, J. Chandrasekhar, G.W. Spitznagel and P.v.R. Schleyer, /. Comp. Chem., 1983,4, 294-301. [ Links ]
38 M.J. Frisch, J.A. Pople and J.S. Binkley, /. Chem. Phys., 1984, 80, 3265-3269. [ Links ]
39 R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454-464. [ Links ]
40 M.E. Casida, C. Jamorski, K.C. Casida and D.R. Salahub, /. Chem. Phys., 1998, 108, 4439-4449. [ Links ]
41 R.E. Stratmann, G.E. ScuseriaandM.J. Frisch,/. Chem. Phys., 1998,109, 8218-8224. [ Links ]
42 C. Van Caillie and R.D. Amos, Chem. Phys. Lett., 1999, 308, 249-255. [ Links ]
43 C. Van Caillie and R.D. Amos, Chem. Phys. Lett., 2000, 317, 159-164. [ Links ]
44 F. Furche and R. Ahlrichs, /. Chem. Phys., 2002, 117, 7433-7447. [ Links ]
45 G. Scalmani, M.J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, /. Chem. Phys., 2006,124, 094107: 1-15. [ Links ]
46 S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117-129. [ Links ]
47 S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239-245. [ Links ]
48 V.W.W. Yam, K.K. Tam, M.C. Cheng, S.M. Peng and Y. Wang, /. Chem. Soc., Dalton Trans., 1992,1717-1723; [ Links ] J.R. Winkler and H.B. Gray, Inorg. Chem., 1985, 24, 346-355; [ Links ] V.W.W. Yam, Y.L. Yui, K.M.C. Wong and K.K. Cheung, Inorg. Chim. Acta, 2000, 300, 721-732. [ Links ]
49 F. Refosco, F. Tisato, C. Bolzati and G. Bandoli, /. Chem. Soc., Dalton Trans., 1993, 605-618, and references therein. [ Links ]
50 T. Gerber, D. Luzipo and P. Mayer, /. Coord. Chem., 2005, 58, 637-641, and references therein. [ Links ]
51 P.D. Benny, C.L. Barnes, P.M. Piekarsky, J.D. Lydon and S.S. Jurisson, Inorg. Chem., 2003,42, 6519-6527. [ Links ]
52 T.I.A. Gerber, D. Luzipo and P. Mayer, /. Coord. Chem., 2006, 59, 1515-1519. [ Links ]
53 I. Booysen, T.I.A. Gerber, E. Hosten and P. Mayer, Bull. Chem. Soc. Ethiop., 2008, 22, 101-105. [ Links ]
54 I. Booysen, T.I.A. Gerber, P. Mayer and H.J. Schalekamp, /. Coord. Chem., 2007, 60, 1755-1761. [ Links ]
Received 22 April 2014
Revised 5 October 2014
Accepted 6 October 2014
* To whom correspondence should be addressed. E-mails: thomas.gerber@nmmu.ac.za (TIAG) and aviye@boun.edu.tr (VA)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}