










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban Jan. 2014
RESEARCH ARTICLE
Quininium Malates: partial chiral discrimination via diastereomeric salt formation
Nikoletta B. Báthori*; Ayesha Jacobs; Luigi R. Nassimbeni; Baganetsi K. Sebogisi
Crystal Engineering Research Unit, Department of Chemistry, Cape Peninsula University of Technology, P.O. Box 652, Cape Town, 8000, South Africa
ABSTRACT
Quinine was employed as a resolving agent for racemic malic acid. The resultant product was a quininium salt containing 75 % of the D-malate anion. Quinine was also crystallized with pure L- and D-malic acids and the structures of the resulting diastereomeric salts were elucidated. The crystal packings were analyzed in terms of their non-bonded interactions and the conformation of the quinine, which was compared with other quinine structures recorded in the Cambridge Structural Database. The results indicate that the mechanism of enantiomeric resolution is reliant upon hydrogen bonded interactions.
Keywords: Chiral discrimination, diastereomeric salt, quinine.
1. Introduction
Selectivity arises from the phenomenon of molecular recognition and depends on such factors as complementarity of binding sites, the strengths of the relevant non-bonding interactions and the conformational adaptability of the host molecule.
The most demanding form of separation is that of enantiomeric resolution. This is because the physical properties of the (R)-and (S)-forms of the two optical isomers have identical properties such as melting point, boiling point, vapour pressure, density and differ only in their reactions toward chiral compounds and polarized light. The enantiomeric purity of a compound is important in the analytical, food, pesticide and particularly in the pharmaceutical industries. Resolution of racemic modifications of organic molecules is of considerable importance to the pharmaceutical industry hence these compounds represents close to ca. 30 % of all drug sales worldwide.1 The global pharma market in 2012 was 990 billion USD and a recent estimate of the sale of single-enantiomer drugs is of the order of 300 billion USD.2 When a compound is produced as a racemic modification in certain cases only one of the enantiomers exhibits the required biological effect. Different techniques are used to separate the enantiomers, such as spontaneous resolution by crystallization, enzymatic transformations, and chromatographic methods. Diastereomeric salt formation is also a well-known and widely used technique and a typical method employed in the separation of racemic modifications of carboxylic acids and amines.3
Quinine, the major component of the cinchona alkaloids has a long history as a resolving agent. Pasteur employed its derivative quinicine to resolve tartaric acid.4 Kozma's review of resolutions via diastereomeric salt formation catalogues 352 resolution experiments with quinine, mostly of carboxylic acids, but also lists ketones and alcohols.5 However, relatively few crystal structures of diastereomeric salts have been reported as a recent Cambridge Structural Database6 (CSD, version 5.35, 2014 May) search conducted with ConQuest7. Two of the publications discuss a crystal structure, which contains quinine in a neutral form8 while the majority of the work describes crystal structures in which the quinine moiety is protonated. The salt formation ability of quinine was investigated by exposing quinine to achiral compounds, such as salicylic acid and saccharin.9 In addition a series of chiral diastereomeric salts of quinine were synthesized and their structures were discussed, such as diquinine biphenyl-2,2'-dicarboxylic acid monohydrate (CSD code JIFYOM).10 Quinine was used also to assign the absolute configuration for the building block of a b-peptide oligomer, trans-2-aminocyclohexanecarboxylic acid (LIVWOC11) and a series of products tested to inhibit renal dipeptidase ((-) quininium ( + )-2,2-dimethylcyclopropanecarboxylate monohydrate, FIJSUM1212). Surprisingly, very few structural data are available from resolution experiments conducted with quinine. A racemic modification of [fluoro(hydroxyphenylphosphinyl)methyl]-phosphonic acid was crystallized with quinine and the 98 % diastereomerically pure diquinine salt was obtained (YUKMOG13). Quinine was used also to resolve (±)-hydroazulenone, the starting material for the total synthesis of sesquiterpenoids (FEQZEG14), enriching enantiomers during the preparation of paracyclophen phosphine ligands (BEYTIJ15) and resolving 1,1'-binaphtyl-2,2'-dicarboxylic acid via diastereomeric salt formation (URIJAV16).
A study of chiral discrimination of mandelic acid by quinine was investigated via a series of competition experiments, in which the mole fraction of (R)-mandelic acid was varied systematically from 0 to 1. The experiment yielded a selectivity curve. The concomitant structures of the quininium mandelates were elucidated and it was concluded that the asymmetric unit containing (R)-, (R)- and (S)-mandelate salt of quinine dominates over a wide range of the selectivity curve.17). The naturally occurring isomer of malic acid is the L(+) form and is found in apples and many other fruits and plants. Its metabolism is well understood, but little is known about the fate of D-(-)-malic acid in the body.18 Attempted separation of the two forms was carried out previously by exposing the racemic malic acid to L-citrulline (SACSOE19) and R-phenylethylamine (EYOBAV20).
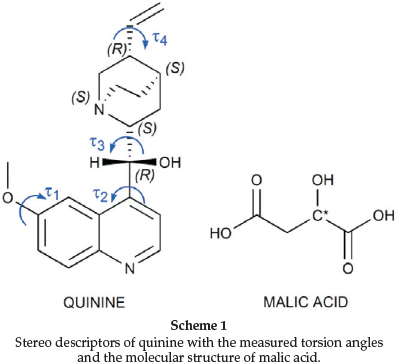
We now present the structures of the salts, which arise, from the combination of quinine (QUIN) with L, D and racemic (DL) malic acid (MA). Four structures were elucidated, namely 1: 2(QUIN+) · (L-MA2-) · 2H2O, 2: 2(QUIN+) · (D-MA2-) · 2H2O, 3:(QUIN+) · (D-MA-) · H2O and 4:2(QUIN+) · ((0.75 D-MA2-) · (0.25 L-MA2-)) · 2H2O (Scheme 1).
2. Experimental Details
2.1. Crystal Growth
All chemicals were purchased from commercial sources (Sigma-Aldrich) and used without further purification. Compounds 1, 2 and 4 were prepared by mixing quinine (100 mg, 0.3 mmol) with the appropriate acid (20 mg, 0.15 mmol) in the stoichiometric ratio of 2:1 using 96 % ethanol as solvent. Needleshaped crystals were obtained by slow evaporation after one week. Compound 3 was prepared by mixing quinine with D-malic acid in 2:1 ratio in water. Slow evaporation yielded block like crystals after a week.
2.2. Structure Analysis
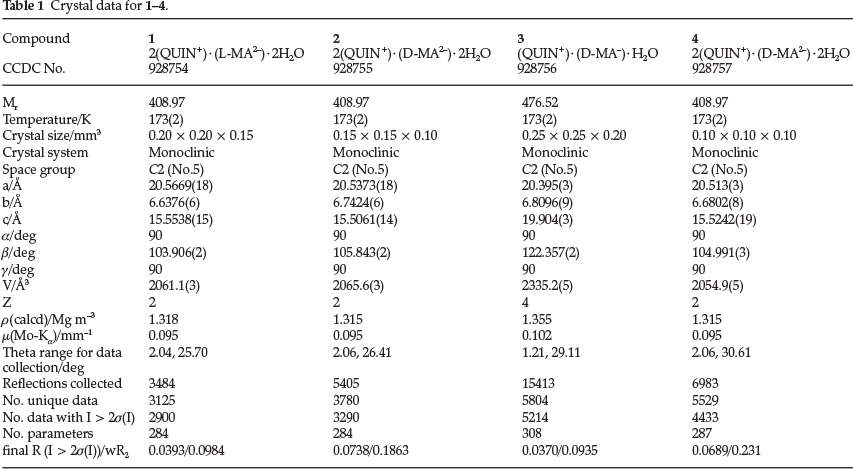
Intensity data of a selected single crystal was collected on a Bruker DUO APEX II diffractometer21 with graphite mono-chromated Mo-Ka1 radiation (l = 0.71073 A) at 173 K using an Oxford Cryostream 700. Data reduction and cell refinement were performed using SAINT-Plus.22 The space group was determined from systematic absences by XPREP23 The structure was solved using SHELXS-9724 and refined using full matrix least squares methods in SHELXL-9721 with the aid of the program X-Seed.25 The hydrogen atoms bound to carbon atoms were placed at idealized positions and refined as riding atoms with Uiso (H) = 1.2 Ueq (Ar-H, CH2) or 1.5 Ueq (CH3). The refinement of the hydroxyl hydrogen and hydrogens of the nitrogen atoms was carried out by first locating them on a difference electron density map and subsequently imposing an appropriate bond length constraint. Diagrams and publication material were generated using PLATON,26 X-Seed and Mercury (3.1).27 Crystal data and structure refinement parameters are given in Table 1. CCDC 928754-928757 contain the supplementary crystallographic data for structures 1-4. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, by e-mailing data_requestccdc.cam.ac.uk, or by contacting the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44(0)1223-336033.
3. Results
3.1. Structures
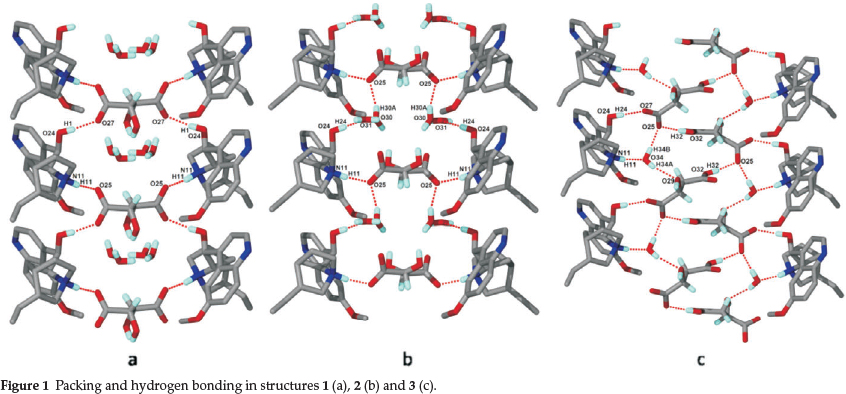
Compound 1 crystallizes in the space group C2 with Z = 2. The chiral carbon C9 of quinine is known to have (R)-configuration and it was assigned accordingly. Protons from each carboxylic acid moiety have been transferred to the quinuclidine nitrogens (N11) of quinine, yielding a salt of stoichiometry 2(QUIN+) · (L-MA2-) · 2H2O. The malate anion is located on a diad at Wyckoff position b and therefore the hydroxyl group has site occupancy of 0.5. The packing of the quininium cations, the malate anions and the disordered water molecules are shown in Fig. 1a. The hydrogen bond structure forms a ring comprising (QUIN)NH+...-OOC(L-MA)COO-... + HN(QUIN)OH... -OOC(L-MA)COO- and may be described with the graph set28 as IMAGEMAQUI(25). The metrics of the hydrogen bonding for this and the other structures are given in Table 2.
Structure 2 is similar to 1, in that it also crystallizes in C2 with Z = 2 and the malate anion is again located on a diad and displays similar hydrogen bonds. The only difference occurs in the packing of 2 which is displayed in Fig. 1b. The hydrogen bonding forms a large ring structure comprising (QUIN)NH+... -ooc(d-ma)coo-...+hn(quin)oh...h2o...-ooc(d-ma) COO...H2O... HO(QUIN+). The water is disordered over two positions and modelled with half site occupancies. The hydrogen bond pattern may be described as IMAGEMAQUI (30).
Structure 3 has a different stoichiometry in that the cation:an-ion ratio is 1:1 and this may be explained by the crystallization conditions. Quinine solubility is very good in alcohols (1 g dissolves in 0.8 mL alcohol29) but drastically less in water (1 g dissolves in 1900 mL water). Malic acid solubility is only slightly higher in alcohols (86.60 g 100 g-1 ethanol) than in water (36.35 g 100 g-1 water). Therefore changing the crystallization solvent from ethanol to water has significant effect on the stoichiometric ratio of the crystal. The structure again crystallizes in C2 with Z = 4 and all moieties lie in general positions. The water is ordered and bridging between the quininium and the malate moieties (Fig. 1c). The hydrogen bonding comprises a ring (QUIN)NH+... H2O...-OOC(D-MA)COOH...-OOC(D-MA)COOH...-OOC(D-MA)...HO(QUIN+) and can be described as Rf(25).
Structure 4 was obtained by exposing QUIN to DL-malic acid. The QUIN partially resolved the racemic mixture and the resultant crystals yielded the malate salt with D:L in the ratio of 75:25. Therefore compound 4 has the formula 2(QUIN+) · ((0.75 D-MA^) · (0.25 L-MA^)) · 2H2O. The structure is similar to 2, with the exception of the water of crystallization being ordered. The hydrogen bonding with the major component is as described for 2.
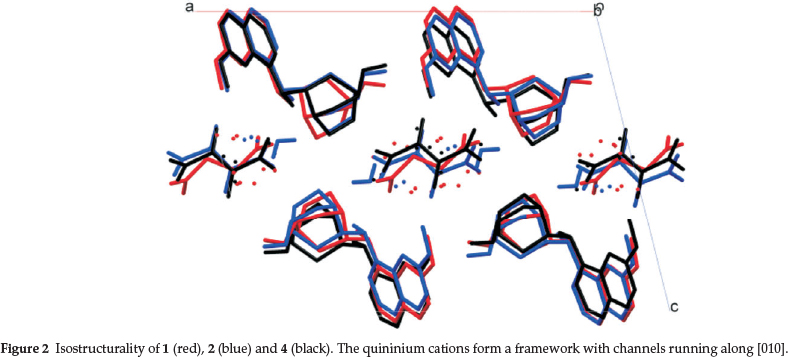
Structures 1, 2 and 4 are essentially isostructural, in that the quininium cations form a framework with channels which contain diads running along [010]. These channels accommodate the malate anions which are therefore disordered and also some water molecules. In structures 1 and 2 different positions are adopted by the L- or D-malate anions, but they share a common position for the O atom that is a hydrogen bond acceptor from the quininium cation. (Fig. 2.) The nature of the disorder in 4 is clear hence the channels are inherently 2-fold symmetric.
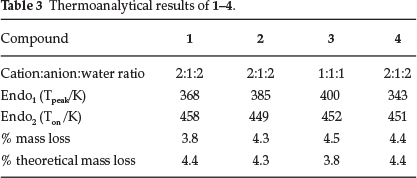
3.2. Thermoanalysis
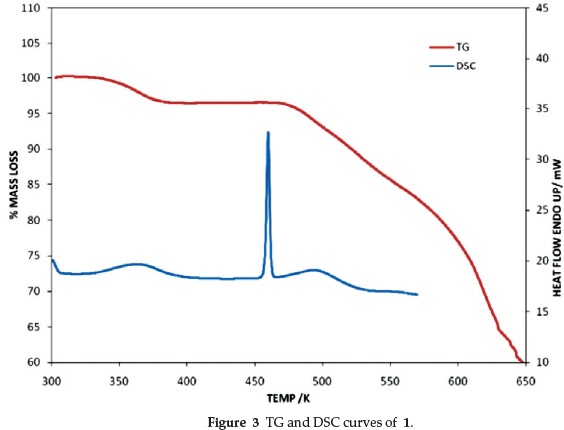
The thermogravimetric (TG) and the differential scanning calorimetry (DSC) curves for the four compounds are very similar, therefore we show the results of 1 as being representative (Fig. 3). For all compounds, the onset and peak temperatures as well as the theoretical and measured mass losses are reported in Table 3. The TG curve shows an initial mass loss attributed to the loss of water of crystallization. The curve is then flat to ca. 470 K after which the salt decomposes. The concomitant DSC curve exhibits an initial endotherm corresponding to the loss of H2O, followed by a sharp peak due to the melt of the salt. There is good agreement between experimental and theoretical mass losses. Because the loss of water gives rise to a broad endotherm, we report the peak and not the onset temperatures.
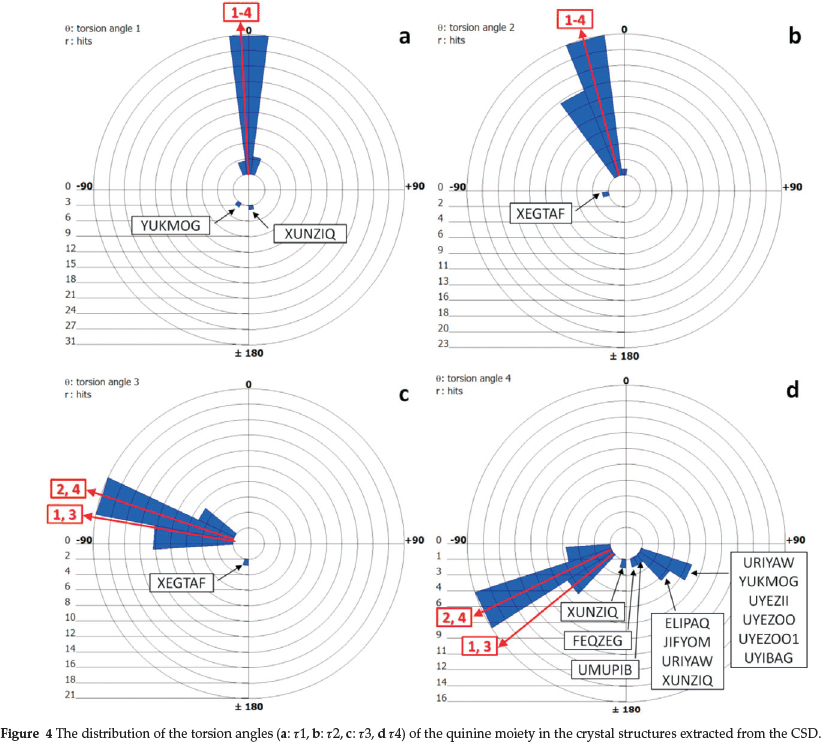
However, some structures do not fit the trend. In the structures of YUKMOG (diquininium (fluoro(hydroxyphenyl-phosphinyl)-methyl)phosphonate acetone solvate sesquihy-drate30) and XUNZIQ (6',9-dihydroxycinchonan-1,1'-diium hexachlorobismuthate chloride monohydrate31) there are two symmetry independent quininium cations in the asymmetric unit and in one of them the methoxy group related to r1 occupies the conformation at t ~180 ° (Fig. 4a).
3.3. Conformation Analysis
We have recorded the four torsion angles r1 (C5-C6-O22-C23), t2 (C3-C4-C9-O24), r3 (024-C9-C10-N11) and r4 (C14-C13-C18-C19) which define the conformation of quinine. (Scheme 1, Table 4) We have compared our findings to the search conducted on the published quinine structures in the CSD. The torsion angles of the 26 structures (overall 40 hits) are shown in Table 5. The polar histograms of the search results and the torsion angles of the four currently reported structures are shown in Fig. 4. We note that t1, r2 and r3 are remarkably constant. The torsion angles of the four new structures are marked as red arrows on the perimeters and they fit the trends shown on the polar histograms.
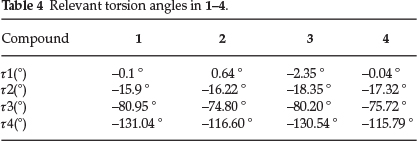
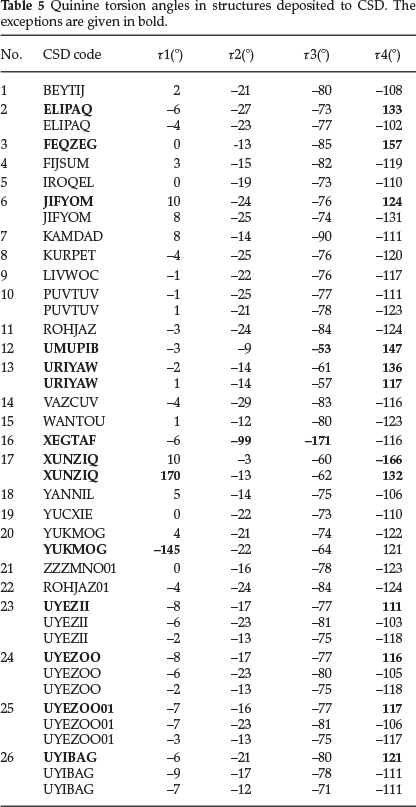
The distribution of r2 shows one structure (XEGTAF: b-cyclo-dextrin (6-methoxyquinolin-4-yl) (5-vinyl-1-azabicyclo[2.2.2] oct-2-yl)methanol clathrate hydrate32) where the hydroxyl group of the quinine is nearly perpendicular to the quinoline ring. (Fig. 4b) Also XEGTAF shows significant difference in t3. (Fig. 4c) It is interesting to note that in this structure the quinuclidine moiety is included in a b-cyclodextrin and the authors suggested that this is the reason for its exceptional conformation. The distribution of r4 suggests that in the majority of the structures the torsion angle of the ethylene moiety centred around -118 °. However, 13 structures show remarkable differences. 12 are clustered between 111-157 ° and in one (XUNZIQ) t4 is -166 °. (Fig. 4d) It is noteworthy that the flexibility of the ethylene moiety has been connected to the chiral selectivity of quinine in diastereomeric salt formation with mandelic acid (UYEZII, UYEZOO, UYEZOO1 and UYIBAG17) These diastereo-meric pairs do not show any significant difference in any of the four torsion angles but it was concluded that the resolution of mandelic acid by quinine is driven by intermolecular hydrogen bonds and slight concomitant changes in the r4 torsion angle.
3.4. Hirshfeld Surface Analysis
The precise mechanism of chiral discrimination is still unknown. Very few solid state studies have been carried out to date; however, there are some current attempts to investigate the process in solution via combining theoretical and experimental studies.34 In agreement all these works point out the importance of the non-bonded interactions, particularly the hydrogen bonds in the discrimination process.
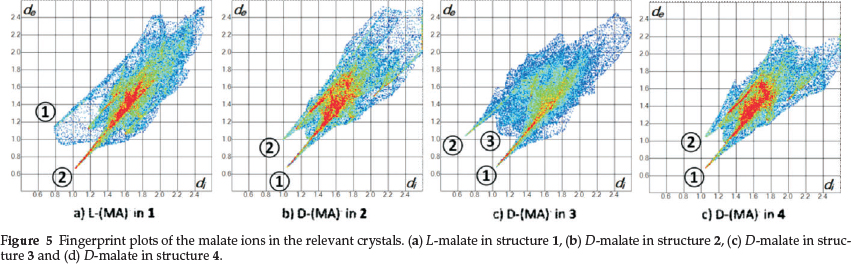
In order to understand the mechanism of resolution of the DL-malic acid by QUIN, we analyzed the non-bonded interactions which occur in the hydrated (QUIN+)(MA-)· salts. We employed the program Crystal Explorer35 which calculates the Hirshfeld surfaces of a molecule or ion in a crystal structure and depicts all the molecular interactions of the targeted moiety with its neighbours in the form of fingerprint plots.
In Fig. 5a the fingerprint plots for the malate anion is shown. The spike labelled © is associated with the hydrogen bonds (QUIN + )N11-H11...O25(L-MA-) and (QUIN + )-OH... O27(L-MA-) which contribute 58 % to the overall interactions towards the procrystal. The spike labelled IMAGEMAQUI is due to (L-MA-)O29- H29...O30/O31 (disordered water). The area between these two spikes is populated by H...H interactions (28 %).
In Fig. 5b, spike © is derived from a combination of (QUIN+) N11-H11...O25(D-MA-), the disordered water O30-H30...025(D-MA-), and (QUIN+)C12-H12B...027(D-MA-) with a total contribution of 62%. Spike © is due to H...H interactions between MA- and the methoxy hydrogens on QUIN+ (27 %).
The fingerprint of the malate anion in 3 is distinctly different and exhibits two symmetrical sharp and a broad spike between them (Fig. 5c). © is associated with (D-MA-)032-H32... 025(D-MA-) and (water) 034-H34B...025(D-MA-) and contribute to the overall interactions with 53 %. © is due to (D-MA-) 032-H32...025(D-MA-) and (D-MA-)029-H29... 027(D-MA-) (9.6 %). Spike © is due to H...H interactions (31.7 %).
The major component of the malate anion in structure 4 is very similar to that of 2, the only difference being that in 4 the water of crystallization is ordered. The fingerprint pattern of the major component of 4 resembles that of 2 closely and is distinct from that of 1.
The resolution experiment with quinine yielded the D-malate salt as the major component and one notes that the hydrogen bonding in this structure is more favourable as it involves the water of crystallization while in 1 the water is not hydrogen bonded. (Fig. 1a and Fig. 1b) This is indeed reflected in the difference and % contributions exhibited by the fingerprint plots.
4. Conclusions
The crystallization of quinine with racemic malic acid yielded the crystals of the D-malate salt as the major component with the QUiN:MA ratio of 1:0.5 (structure 4). The structures of the quininium-L- and D-malate presented the same quininium: malate ratio (structures 1 and 2, respectively). In structure 3, quinine crystallizes with D-malic acid with a different stoichiometry, the QUIN:MA ratio is 1:1. The conformation of the quinine moiety in each structure was compared to the 26 known structures of quinine obtained from the CSD and showed no significant differences. The D-malate is the major product because its structure has a more favorable hydrogen bond pattern.
Supplementary Material
Details to download the X-ray data of the four compounds are provided in the experimental section.
Acknowledgements
Authors thank for Prof. Andrew D. Bond for his valuable comments on the manuscript and the National Research Foundation (Pretoria) and the Cape Peninsula University of Technology for financial support. The authors declare that they have no conflict of interest.
References
1 S.C. Stinson, CENEAR, 2000, 78, 55-78. [ Links ]
2 (a) R.M. Kellogg, J.W Nieuwenhuijzen, K. Pouwer, T.R. Vries, Q.B. Broxterman, R.F.P. Grimbergen, B. Kaptein, R. M. La Crois, E. de Wever, K. Zwaagstra and A. C. van der Laan, Synthesis, 2003, 10, 1626-1638 (b) Facts and Figures, Int. Fed. Pharm. Man. & Assoc., 2012, http://www.ifpma.org/ [ Links ]
3 (a) S.H. Wilen(1971) Resolving Agents and Resolution in Organic Chemistry - Topics in Stereochemistry, Wiley-Interscience, New York, USA, 1971,6,107-176. [ Links ] (b) J. Jacques, A. Collet, S.H. Wilen, Enantiomers, Racemates and Resolutions, Krieger Publishing Company Malabar, FL., 1991 (c) F Toda, [ Links ] ed., Enantiomer Separation. Fundamentals and Practical Methods, Kluwer Norwell, MA. 2004. [ Links ]
4 L. Pasteur, C. R. Acad. Sci., 1853, 37, 162-166. [ Links ]
5 D. Kozma, ed., Optical Resolutions via Diastereomeric Salt Formation, CRC Press, New York, 2002. [ Links ]
6 FH. Allen, Acta Cryst, 2002, B58, 380-388. [ Links ]
7 I.J. Bruno, J.C. Cole, PR. Edgington, M. Kessler, C.F Macrae, P McCabe, J. Pearson and R. Taylor, Acta Cryst., 2002, B58, 389-397. [ Links ]
8 (a) B. Pniewska and A. Suszko-Purzycka, Acta Cryst., 1989, C45, 638-642. [ Links ] (b) G. Paliwoda, B.J. Oleksyn, J. Sliwinski, Enantiomer, 2002, 7, 387-396. [ Links ]
9 (a) B. Oleksyn and P. Serda, Acta Cryst., 1993, B49, 530-535. [ Links ] (b) P. M. Bhatt, N.V. Ravindra, R. Banerjee and G.R. Desiraju, Chem. Commun., 2005, 1073-1075. [ Links ]
10 (a) P.J.F. Griffiths, Acta Cryst., 1959, 12, 418-419. [ Links ] (b) PM. Kimpede and L. Van Meervelt, Acta Cryst., 2010, E66, o2443-o2444. [ Links ] (c) M. Kubicki, T. Borowiak, M. Gawron, M. Giel, J. Gawronski, J. Crystallogr. Spectrosc. Res., 1990, 20, 447-455. [ Links ] (d) A. Gjerlov and S. Larsen, Acta Cryst., 1997, C53, 1505-1508. [ Links ] (e) C. Ryttersgaard and S. Larsen, Acta Cryst., 1998, C54, 1698-1701. [ Links ] (f) C. Ryttersgaard and S. Larsen, Acta Cryst., 2004, E60, o4-o5. [ Links ] (g) B. Samas, A. C. Blackburn and K. C. Kreuchauf, Acta Cryst., 2005, E61, o3983-o3984. [ Links ]
11 D.H. Appella, L.A. Christianson, I.L. Karle, D.R. Powell, S.H. Gellman, J. Am. Chem. Soc., 1999,121, 6206-6212. [ Links ]
12 D.W. Graham, W.T. Ashton, L. Barash, J.E. Brown, R.D. Brown, L.F Canning, A. Chen, J.P Springer and E.F. Rogers, J. Med. Chem., 1987, 30, 1074-1090. [ Links ]
13 R. Bau, P-T.T. Pham, G.D. Duncan, C.E. McKenna, J. Med. Chem., 1995, 38, 1575-1578. [ Links ]
14 W. Tochtermann, T. Panitzsch, M. Petroll, T. Habeck, A. Schlenger, C. Wolff, E.-M. Peters, K. Peters, H.G. von Schnering, Eur. J.Org. Chem., 1998, 2651-2657. [ Links ]
15 B. Dominguez, A. Zanotti-Gerosa, W. Hems, Org. Lett., 2004, 12, 1927-1930. [ Links ]
16 A. Jacobs, L.R. Nassimbeni, A. Sayed and E. Weber, J. Chem. Cryst., 2011,41, 854-857. [ Links ]
17 N.B. Báthori, L.R. Nassimbeni and C.L. Oliver, Chem. Commun., 2011, 47, 2670-2672. [ Links ]
18 FAO Nutrition Meetings Report Series, No. 40, A, B, C, WHOLE/Food Ad./67.89 [ Links ]
19 P.P. Toffoli, N. Rodier, R. Ceolin, F. Lepage, J. Astoin, Acta Cryst., 1988, C44, 2128-2131. [ Links ]
20 D.E.Turkington, G. Ferguson, A.J. Lough, C. Glidewell, Acta Cryst., 2004, C60, o617-o622. [ Links ]
21 Bruker 2005. APEX2. Version 1.0-27. Bruker AXS Inc., Madison, Wisconsin, USA. [ Links ]
22 Bruker 2004. SAINT-Plus (including XPREP). Version 7.12. Bruker AXS Inc., Madison, Wisconsin, USA. [ Links ]
23 Bruker 2003, XPREP2. Version 6.14. Bruker AXS Inc., Madison, Wisconsin, USA. [ Links ]
24 G.M. Sheldrick, SHELXS-97 and SHELXL-97 Programs for crystal structure determination and refinement. University of Göttingen, 1997. [ Links ]
25 L.J. Barbour, J. Supramol. Chem., 2001, 1, 189-191. [ Links ]
26 A.L. Spek,PLATON, AMultipurpose Crystallographic Tool,UtrechtUni-versity, Utrecht, Netherlands, 2008. [ Links ]
27 C.F. Macrae, I.J. Bruno, J.A Chisholm, PR. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek, P.A Wood, J. Appl. Cryst., 2008, 41,466-470. [ Links ]
28 M.C. Etter, J.C. MacDonald, J. Bernstein, Acta Cryst., 1990, B46, 256-262. [ Links ]
29 M.J. O'Neil, ed., The Merck Index - An Encyclopedia of Chemicals, Drugs and Biologicals, 2006, Merck and Co. Inc., Whitehouse Station, NJ, USA. [ Links ]
30 R. Bau, P-T.T. Pham, G.D. Duncan, C.E. McKenna, J. Med. Chem., 1995, 38, 1575-1578. [ Links ]
31 H.L.Cai, T. Zhang, L.Z. Chen, R.G. Xiong, J. Mater. Chem., 2010, 20, 1868-1870. [ Links ]
32 Z. Fan, C.H. Diao, H.B. Song, Z.L. Jing, M. Yu, X. Chen, M.J. Guo, J. Org. Chem. 2006, 71, 1244-1246. [ Links ]
33 (a) N.B. Báthori, L.R. Nassimbeni, Cryst. Growth and Design, 2010, 4, 1782-1787. [ Links ] (b) N.B. Báthori, L.R. Nassimbeni, Cryst. Growth and Design, 2012, 12, 2501-2507. [ Links ] (c) A. Bialonska, Z. Ciunik, Cryst. Growth and Design, 2013,13, 111-120. [ Links ]
34 R. Arjumand, I.I. Ebralidze, M. Ashtari, J. Stryuk, N.M. Cann and J.H. Horton, J. Phys Chem. C, 2013, 117, 4131-4140. [ Links ] (b) X.-H. Zhang, H.-L. Wu, X.-L. Yin, L.-H. Li, J-Y. Wang, Y. Chen, C. Kang, R.-Q. Yu, Anal. Methods, 2013, 5, 710-717. [ Links ]
35 (a) J.J. McKinnon, M.A. Spackman and A.S. Mitchell, Acta Crystallogr., 2004, B60, 627-668; [ Links ] (b) M.A. Spackman and J.J. McKinnon, CrystEngComm, 2002, 4, 378-392. [ Links ]
Received 22 July 2014
Revised 16 September 2014
Accepted 16 September 2014
* To whom correspondence should be addressed. E-mail: bathorincput.ac.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}