










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban Jan. 2014
RESEARCH ARTICLE
Three-component Synthesis of Electron-poor Alkenes using Isatin Derivatives, Acetylenic Esters, Triphenylphosphine and Theoretical Study
Masoome SheikhiI, *; Davood SheikhII; Ali RamazaniIII
IYoung Researchers & Elite Club, Gorgan Branch, Islamic Azad University, Gorgan, Iran
IIYoung Researchers & Elite Club, Hamedan Branch, Islamic Azad University, Hamedan, Iran
IIIDepartment of Chemistry, University of Zanjan, Zanjan, Iran
ABSTRACT
Synthesis of electron-poor alkenes has been reported by 1,2-proton shift and elimination of triphenyl phosphine from phosphorus ylide in good to high yields. The structures of six novel products were deduced from their IR, 1H NMR, and 13C NMR spectra. The B3LYP/HF calculations for computation of 1H and 13C NMR chemical shifts have been carried out for the compounds with the 6-31G* basis set utilizing the GIAO approach. In addition, theoretical configurations of the title compound were studied in terms of the combined analysis of the HOMO-LUMO energy gap, NBO analysis, thermodynamic parameters and molecular electrostatic potential (MEP). Also ionization potential (I), electron affinity (A), chemical hardness (η), electronic chemical potential (μ) and electrophilicity (ω) of the title molecule are reported. All calculations were performed using B3LYP method with the 6-31G* basis set.
Keywords: Isatin, electron-poor alkenes, DFT, NBO, HOMO, LUMO.
1. Introduction
The development of new stereoselective reactions has been a major topic in synthetic organic chemistry.1-3 Organophosphorus compounds have been used in organic synthesis as useful reagents as well as ligands of a number of transition metal catalysts.4
Acetylenic esters are reactive systems taking part in many chemical synthesis, for example, as Michael acceptors.5 In recent years, there has been increasing interest in the application of acetylenic esters for multi-component synthesis.6-9 Multi-component reactions (MCRs) have proved to be notably successful in generating products in a single synthetic operation.10-11 The development of new MCRs and improvement of known multicomponent reactions are the subjects of considerable current interest.
Acrylic compounds are good Michael acceptors due to their great reactivity as electrophiles towards nucleophiles, particularly in the preparation of natural and biologically active com-pounds.12-15 Acrylates form a versatile class of polymers that play a major role in military and commercial products.16 Compounds with alkyl acrylate skeletons have significant importance in material science.17
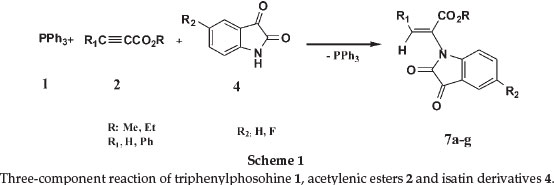
Because of their wide range of industrial and synthetic applications, substituted acrylates have recently received a great deal of attention.18-20 Therefore, in this paper, we present a simple, mild and efficient synthesis of these compounds in good yields (Scheme 1).
In recent years, computational chemistry has become an important tool for chemists and a well-accepted partner for experimental chemistry.21-26 Density functional theory (DFT) and Hartree-Fock (HF) methods have become a major tool in the methodological arsenal of computational organic chemists. Recently, the GIAO (gauge including atomic orbitals) method for the calculation of NMR chemical shifts have been implemented in major quantum chemistry packages.27-29 The GIAO approach facilitates accurate NMR shift calculations via electron-correlated methods. In this paper we will discuss the accuracy of the calculation of 1H and 13C chemical shifts performed at both the HF and density functional theory (B3LYP) frameworks using the GIAO technique with the 6- 31G* basis set.
2. Results and Discussion
On the basis of the well established chemistry of trivalent phosphorus nucleophiles,22,30 it is reasonable to assume that phosphorus ylides 6 result from the initial addition of triphenylphosphine to the acetylenic ester and protonation of the 1:1 adduct by isatin derivatives, then the positively charged ion is attacked by the isatin anion. The scope of the reaction was extended to a variety of structurally diverse acetylenic esters. All the reactions proceeded smoothly to provide the corresponding new products 7a-g in satisfactory yields and the results are summarized in Table 1. Compounds 7a, 7b, 7c, 7e and 7g are stable solid powders. IR, 1H NMR and 13C NMR data are useful information for the structural assignment of the products. We isolated 7a and 7b as a mixure of diastereomers, which were then
separated with column chromotography. The 1H NMR (CDCl3) spectra of the crude product (mixture: 7a and 7b) show the presence of two stereoisomers (7a: E and 7b: Z).31 Also the 1H NMR spectra show that the two stereoisomers (E and Z) are not in equilibrium (E/Z: 46/54). The relative population of E and Z isomers were determined via their 1H NMR spectra (based on reported relative intensities of the olefinic protons of E and Z isomers ( = CH)).31,32 Also, product 7e was isolated as one stereoisomer Z. The 1H NMR spectrum of 7a, 7b and 7e exhibited one singlet line at 5.29, 8.11 and 8.10 ppm for the =CH group.
Further confirmation of the structures was obtained from the 13C NMR spectra which displayed olefinic carbon (=CH) resonances of 7a, 7b and 7e at about 132.5,131.6 and 131.5, respectively, and carbonyl carbons at about d = 158-183 (see Experimental section).32 The shift at 8.11 ppm of the =CH group of the Z rotamers (7b and 7e) is deshielded. Also the 1H NMR spectrum of 7c, 7d and 7g displayed signals for =CH2 protons as two sets of singlets at d = 6.09, 7.26, d = 7.26, 7.69 and d = 7.54, 7.62 ppm, respectively.
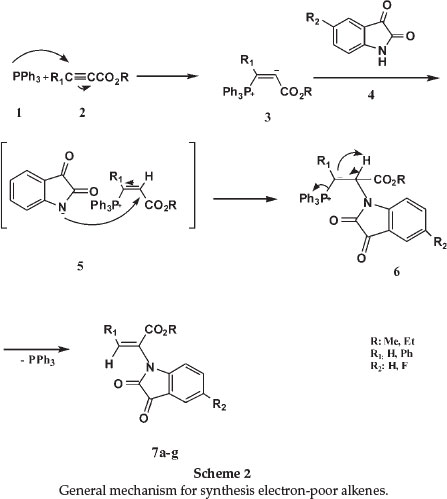
The theoretical results of NMR calculations of the structures correspond with the experimental data (see Theoretical section, Tables 2 and 3). This supports the proposed structures.
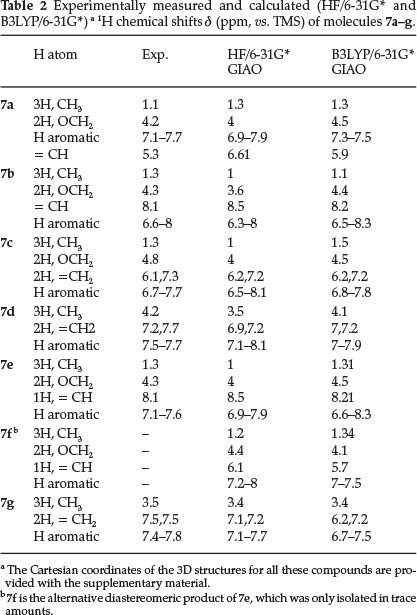

A possible mechanism to explain the formation of the products 7a-g is depicted in Scheme 2. The formation of products 7a-g can be rationalized by initial formation of intermediates 3 through the standard Michael addition of the triphenyl-phosphine 1 to the b-carbon of the electron-deficient alkyne 2. Protonation of 3 by isatin derivatives 4 leads to vinyltriphenyl-phosphonium salts 5, which undergoes a Michael addition reaction with conjugated base to produce phosphorus ylides 6. The ylides are converted to products 7a-g via elimination of triphenylphosphine under the correct reaction conditions.
2.1.1H and 13C NMR Chemical Shifts
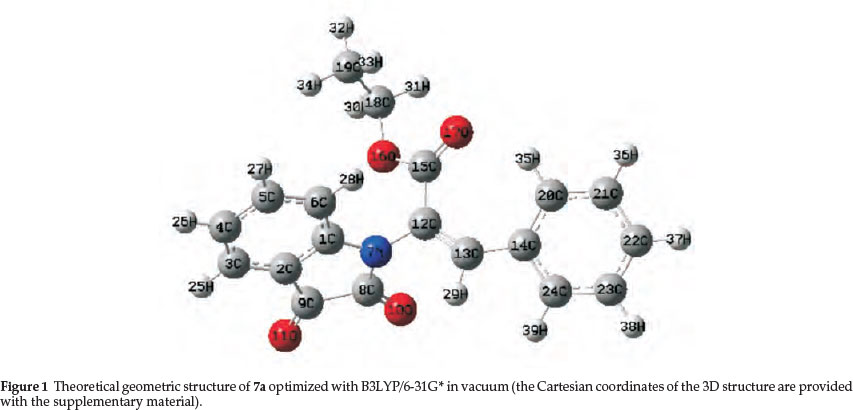
In the present work, we have calculated the 1H and 13C NMR chemical shifts using HF/B3LYP methods with the 6-31G* basis set and the GIAO approach for compounds 7a-g. The theoretical geometric structure of 7a is shown in Fig. 1. The observed 1H and 13C NMR chemical shifts and the calculated values are presented in Tables 2 and 3, respectively.
The theoretical 1H NMR data of 7a, 7b and 7e exhibited one singlet at 5.9, 8.2 and 8.21 ppm for the =CH group (B3LYP/ 6-31G*). Also, the theoretical 13C NMR for the olefinic carbon (=CH) of 7a, 7b and 7e are displayed at about 133.6, 130.8 and 131.4, respectively (B3LYP/6-31G*).
The theoretical calculations were performed for the diastereo-meric alkene of 7e (isomer 7f, which was isolated in trace amounts, see Tables 2 and 3). The theoretical 1H NMR data of 7f exhibited one singlet at 5.7 ppm for the =CH group (B3LYP/6-31G*). Also, the theoretical 13C NMR for the olefinic carbon ( = CH) of this alkene is displayed at about 133.4 (B3LYP/6-31G*, see Tables 2 and 3). Both the proton and 13C NMR deviate considerably from the experimentally observed signals for 7e, confirming the correct assignment thereof.
On the basis of our calculations and experimental 1H and 13C NMR chemical shifts, we observed excellent agreement between the experimental and theoretical results. This confirms our initial NMR assignments for these compounds. In order to compare this agreement, the correlation graphic based on the theoretical and experimental data was investigated. The correlation value (R2) of molecule 7a for HF/6-31G* and B3LYP/6-31G* is 0.99 and 0.9995, respectively (Fig. 2).
2.2. Thermodynamic Analysis
Thermodynamic analysis indicates that the relative energies (ΔΕ), enthalpies (AH) and Gibbs free energy (AG) are negative for molecules 7a-g whereas the calculated entropies (AS) are positive. This indicates that these molecules are stable in the gas phase (see Table 4).
Dipole moment is a good measure for the asymmetric nature of a molecule. The values of dipole moment for molecules 7a-g are listed in Table 5. The size of the dipole moment depends on the composition and dimensionality of the 3D structures. All structures have relative high dipole moment values. All the structures have the C1 point group, which refers to high asymmetry of the structure. As can be seen, the dipole moment for structure of 7b is relatively high (B3LYP/6-31G* = 5.8233 Debye). The high value of dipole moment for 7b is due to its asymmetric character. Since product 7b is the Z isomer, the atoms are irregularly arranged which gives rise to the increased dipole moment (with respect to 7a at 4.94 Debye).
In order to study the atomic charge distribution of compound 7a, the natural bond orbital charges (NBO) have been calculated and are presented in Fig. 3. As seen in Fig. 3, the atomic charge distribution is different. As expected, the O10, On,
O16 and O17 atoms have negative charges (-0.529e, -0.480e, -0.547e and -0.612e, respectively) whereas all the hydrogen atoms carry positive charge. The N7 atom has a large negative charge (-0.469e). All carbon atoms of phenyl ring exhibit negative charges. The C8, C9 and C15 (carbon of carbonyl groups) have more positive charge (0.635e, 0.485e and 0.795e, respectively), as is expected. These data clearly show that ester carbonyl carbon (C15) is the most reactive electrophilic atom.
2.3. NBO Analysis
Natural bond orbital analysis is an important method for studying intra- and inter-molecular bonding and interaction between bonds. The results of natural bond orbital (NBO) analysis and the polarization coefficient values of atoms for molecule 7a are listed in Table 6. According to Table 6, the calculated bonding orbital for the C1-N7bond is BD = 0.6179 sp268 + 0.7862 sp2 04. The polarization coefficients of C1 = 0.6179 and N7 = 0.7862 suggest that N7 is more electron-rich than the C1 atom. The bonding orbital for the N7-C8 bond is BD= 0.8033 sp212 + 0.5956 sp219.
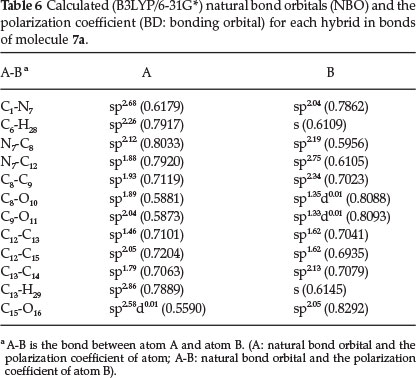
These polarization coefficients (N7 0.8033 and C8 0.5956), also suggest that N7 is more electron-rich than the C8 atom. The calculated natural charge (NBO) of the N7 atom is negative (-0.469e) whereas C8 has a positive value (0.635e). The bonding orbital of the C12-C15 is BD = 0.7204 sp205 + 0.6935 sp162. These polarization coefficient (C12 0.7204 and C15 0.6935) suggest that C12 is more electron-rich. The calculated natural charge (NBO) of C15 atom is more positive (0.795e) than C12 atom (0.029e). Thus more charge density resides on C12. For all the C-O bonds, the polarization coefficient value of oxygen atom is greater than the C atom (see Table 6), as expected.
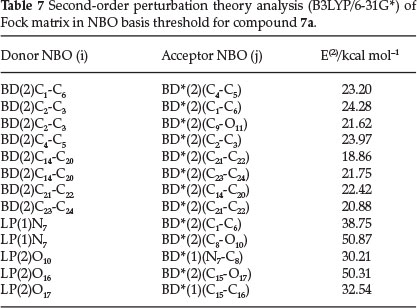
Electron donor orbital, acceptor orbital and the interacting stabilization energy resulting from the second-order micro disturbance theory33,34 are reported in Table 7. The electron delocalization from filled NBOs (donors) to the empty NBOs (acceptors) describes a conjugative electron transfer process between them. For each donor (i) and acceptor (/), the stabilization energy E(2) associated with the delocalization i → j is estimated. The resonance energy (E(2)) shows the amount of participation of electrons in expected the resonance between atoms. According to the results of the NBO analysis for molecule 7a (Table 7), the greatest resonance energy (E(2)) is 50.87 for LP(1)N7 that participates as donor and C8-O10 [anti-bonding BD*(2)] as acceptor. These results suggest that charge is transferred from N7 to C8-O10 (N7 → C8-O10). The calculated natural charge of N7 (-0.469e) and C8 (0.635e) that are taking part in intramolecular charge transfer is indicated in the NBO analysis. The LP(2)O16 participates as donor and the anti-bonding orbital for BD*(2)(C15-O17) acts as acceptor resulting in favorable resonance energy (E(2)) (O16 → C15-O17, 50.31 kcal mol-1). Also BD(2)(C2-C3) participates as donor and the anti-bonding BD*(2)(C1-C6) and BD*(2)(C9-O11) orbital act as acceptor. The resonance energies (E(2)) for the transfer of electron density from BD(2)(C2-C3) of the isatin ring to the anti-bonding BD*(2)(C1-C6) 2and3 BD*(2)(C9-O11) orbitals of the isatin ring are 24.28 and 21.62, respectively. These values indicate large charge transfer from the bonding orbital for BD(2)(C2-C3) to the anti-bonding orbital for BD*(2)(C1-C6) [C2-C3 → C1-C6].
2.4. Electronic Properties
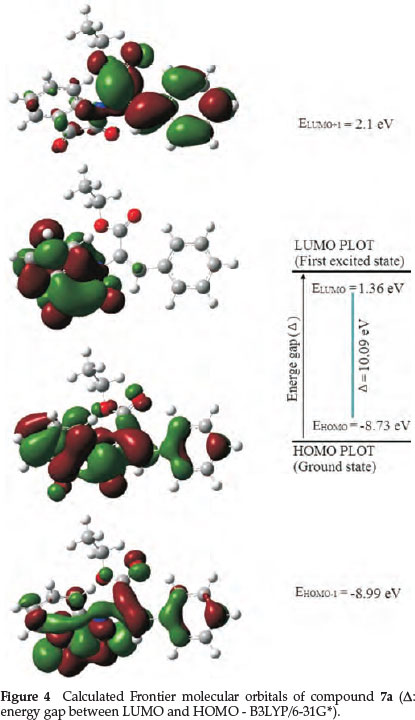
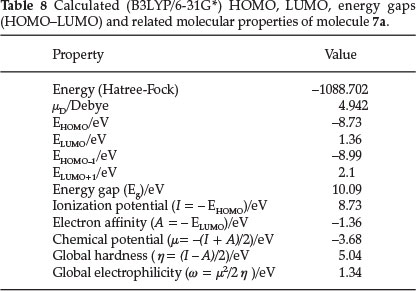
Quantum chemical methods are important to obtain information about molecular structure and electrochemical behaviour. Figure 4 shows the results of the frontier molecular orbitals (FMO) analysis calculated for molecule 7a with B3LYP/6-31G*. According to Fig. 4, charge transfer is taking place within the molecule. The HOMO and HOMO-1 orbitals are localized mainly on the isatin ring and phenyl ring whereas LUMO and LUMO+1 orbitals are localized mainly on the isatin ring. The calculated values are -8.99, -8.73, 1.36, 2.1 and 10.09 eV for EHOMO-1, EHOMO, ELUMO, ELUMO + 1 and the HOMO-LUMO gap (DE), respectively. The energy of the HOMO is directly related to the ionization potential and the energy of the LUMO is directly related to the electron affinity. The HOMO-LUMO gap (DE), is 10.09 eV and such a large energy gap implies high stability for the molecule.3^37 Also ionization potential (I), electron affinity (A), chemical hardness (η), electronic chemical potential (μ) and electrophilicity (w) of molecule 7a were calculated and are listed in Table 8.
3. Experimental
Chemicals used in this work were purchased from Aldrich and Merck chemical companies and used without purification. IR spectra were recorded on a mattson-1000 FT spectrometer using KBr pellets. 1H and 13C NMR spectra were measured for samples in CDCl3 with a BRUKER DRX-250 AVANCE spectrometer at 250 and 62.5 MHz respectively, using Me4Si as internal standard. Melting points were measured on an Electrothermal 9100 apparatus. Elemental analyses for C, H and N were performed using a Perkin-Elmer 2400 series analyzer.
3.1. General Procedure for the Synthesis 7a-g: (note 7f was only observed in trace amounts)
A solution of triphenylphosphine (1.0 mmol, 0.262 g) and isatin derivatives (1.0 mmol) in dry dichloromethane (10 mL) was placed in a three-necked flask in an ice-water bath. Then, a solution of acetylenic esters (1.0 mmol) in dry dichloromethane (4 mL) was added dropwise and stirred for 5 min. After 5 min, the mixture was cooled to room temperature and then stirred for an appropriate time (Table 1) until the reaction was completed as monitored by TLC (n-hexane/EtOAc; 2:1) analysis. The solvent was removed under reduced pressure and the residue was purified by column chromatography with silica gel 60 HF-254 using petroleum ether/EtOAc (10:2) as eluent. All the products were characterized by spectral data (IR, 1H NMR and 13C NMR). The physical and spectral data for the products are given below and the NMR and IR data are also presented with the supplementary material.
Ethyl-(E)-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-prope noate (7a)
Orange crystals (46 %).Mp 168-170 0C. Rf (EtOAc/ n-hexane: 1/2) = 0.34, IR (KBr): n = 3061 (CH, arom), 2984 (CH, alipha), 1746, 1723, 1615 (3C=O), 1469 (C=C, alkene) cm-1,1H NMR ( CDCl3) a (ppm): 1.1 (t, 3H, 3JHH = 7.0 Hz, CH3), 4.2 (q, 2H, 3JHH = 7.0 Hz, OCH2), 7.1-7.7 (m, 9H, arom), 5.3 (s, 1H, =CH). 13C NMR (CDCl3) a (ppm): 14 (CH3), 62.2 (OCH2), 132.5 (=Ch), 111.2, 118.2, 120, 122.5.123.2.124.4.125.128.3.129.6.129.8.138.6.141.2.151.2 (12C of aromatic and =C-N), 158 (C=O of amide), 163 (C=O of ester), 182.4 (C=O of ketone); Anal. Calcd. for C19H15O4N: C, 71.02, H, 4.70, N, 4.36; found C, 70.96, H, 4.81, N, 5.05.
Ethyl-(Z)-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2-prope noate (7b).
Red crystals (54 %). Mp 168-170 0C. Rf (EtOAc/n-hexane: 1/2) = 0.33, IR (KBr): n= 3091 (CH, arom), 2923 (CH, alipha), 1746,1746, 1615 (3C=O), 1469 (C=C, alkene) cm-1, 1H NMR (CDCl3) a (ppm): 1.3 (t, 3H, 3JHH = 7.0, CH3),4.3 (q, 2H, 3JHH = 7.0 Hz, OCH2), 6.6-8 (m, 9H, arom), 8.1 (s, 1H, = CH). 13C NMR (CDCl3) a (ppm): 14.2 (CH3), 62.3 (OCH2), 131.6 (= CH), 111.5,118.2,1:21.1,124.4, 125.8.128.2.129.2.129.8.131.2.135.8.138.7.142.6.150.6 (12C ofar-omatic and =C-N), 158 (C=O of amide), 163.00 (C=O of ester), 181.9 (C=O) of ketone); Anal. Calcd. for C19H15O4N: C, 71.02, H, 4.70, N, 4.36; found C, 71.00, H, 4.92, N, 5.12.
Ethyl-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-acrylate (7c)
Orange solid (85 %). Mp 110 0C. Rf (EtOAc/ n-hexane: 1/2) = 0.35, IR (KBr): n= 3092 (CH, arom), 2923 (CH, alipha), 1746,1730, 1646 (3C=o), 1615 (C=C, alkene) cm-1,1H NMR (CDCl3) a (ppm): 1.3 (t, 3H, 3Jhh = 7.0 Hz, CH3), 4.8 (q, 2H, 3JHH = 7.0 Hz, OCH2), 6.7-7.7 (m, 4H, arom), 6.1,7.3 (2s, 2H, 3J = 0 Hz, = CH2), 13C NMR (CDCl3) a (ppm): 14.1(CH3), 62.3 (OCH2), 128.6 (=CH2), 111.3, 118, 124.3, 125.6, 130.9, 132, 138.4 (6C of aromatic and =C-N), 161.6 (C = O of amide), 161.7 (C = O of ester), 183.4 (C = O of ketone); Anal. Calcd. for C13H11O4N: C, 63.67, H, 5.52, N, 5.71; found C, 63.51, H, 5.74, N, 5.90.
Methyl-2-(2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-acrylate (7d)
Viscouse yellow oil (80 %). Rf (EtOAc/ n-hexane: 1/2) = 0.36, IR (KBr): v = 3084 (CH, arom), 2923 (CH, alipha), 1738, 1707, 1630 (3C=O), 1592 (C=C, alkene)cm-, 1H NMR (CDCl3) d (ppm): 4.2(s, 3H,3Jhh = 0 Hz, CH3), 7.5-7.7 (m, 4H, arom), 7.2,7.7 (2s, 2H, JHH =
0 Hz, =CH2), 13C NMR (CDCl3) d (ppm): 51(CH3), 128.3 (=CH2), 112,125.7,128.6,132,132.2,133.4,145 (6C ofaromatic and =C-N), 159 (C=O of amide), 162 (C=O of ester), 181 (C=O of ketone); Anal. Calcd. for C12H9O4N: C, 62.34, H, 3.92, N, 6.06; found C, 62.13, H, 3.85, N, 5.94.
Ethyl-(Z)-2-(5-fluoro-2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl -2-propenoate (7e)
Red solid (90 %). Mp 1320C. Rf (EtOAc/n-hexane: 1/2) = 0.30, IR (KBr): v= 3069 (CH, arom), 2953 (CH, alipha), 1748, 1730, 1623 (3C=O), 1492 (C=C, alkene) cm-1,1H NMR (CDCl3) d (ppm): 1.3 (t, 3H, 3Jhh = 7.0 Hz, CH3), 4.3 (q, 2H, 3JHH = 7.0 Hz, OCH2), 7.1-7.6 (m, 9H, arom), 8.1 (s, 1H, =Ch), 13C NMR (CDCl3) d (ppm): 14.2 (CH3), 62.3 (OCH2), 131.5 (=CH), 112.8, 118.2, 1211.0, 125, 125.4, 128,3129, 129.2, 1229.8, 131, 131.3, 142.7, 146.6 (12C of aromatic and =C-N), 162.8 (C=O of amide), 162.8 (C=O of ester), 181 (C=O of ketone); Anal. Calcd. for C19H14O4NF: C, 67.25, H, 4.16, N, 4.13; found C, 67.31, H, 4.25, N, 4.08.
Methyl-2-(5-fluoro-2,3-dioxo-2,3-dihydro-1H-indol-1-yl)-3-phenyl-2 -propenoate (7g)
Yellow solid (95 %). Mp 135 0C. Rf (EtOAc/n-hexane: 1/2) = 0.31, IR (KBr): v = 3053 (CH, arom), 2961 (CH, alipha), 1738,1707,1630 (3C=o), 1584 (C=C, alkene) cm-1,1H NMR (CDCl3) d (ppm): 3.5 (s, 3H, 3Jhh = 0 Hz, CH3), 7.4-7.8 (m, 3H, arom), 7.5, 7.6 (d, 2H, 3Jhh = 1.5> Hz, = CH2), 13C NMR (CDCl3) d (ppm): 52 (cH3), 128.4 (=CH2), 112,131.9, 132.2, 132.3,133.5,142.4, 150 (6C ofaromatic and =C-N), 158 (C=O of amide), 163.0 (C=O of ester), 181.9 (C=O of ketone); Anal. Calcd. for C12H8O4NF: C, 57.83, H, 3.23, N, 5.62; found C, 57.93, H, 3.11, N, 5.49).
4. Computational Details
The ab initio molecular calculations were carried out using the Gaussian 98 software package38 at the HF and DFT/B3LYP39 levels of theory with the 6-31G* basis set. Also, geometry optimization and NBO analysis40 in vacuum (gas phase) for molecules were performed. The Cartesian coordinates of the 3D structures for all these compounds are provided with the supplementary material. The 1H and 13C NMR chemical shifts of the compounds were calculated using the GIAO approach. The calculations also provide valuable information for exploring the thermodynamic parameters. We obtained the energy (DE), enthalpies (DH), Gibss free energy (DG), entropies (DS) and constant volume molar heat capacity (Cv) values of products.41-43 Electronic properties such as energy of the highest occupied molecular orbital (EHOMO), energy of the lowest unoccupied molecular orbital (ELUMO), HOMO-LUMO energy gap (Eg), atomic charges and dipole moment (μ) were determined. The optimized molecular structure (Fig. 2), HOMO and LUMO surfaces were visualized using GaussView 03 program.44
5. Conclusions
We have described a convenient three-component route for the preparation of six novel electron-poor alkenes 7a-g. The reactions were carried out in one-pot without separation and purification of the intermediates 3,5 and 6. The present procedure has many advantages such as good to high yields, environmentally friendly nature and fairly mild reaction conditions. In the present study also, 1H and 13C NMR chemical shifts were obtained using theoretical calculations. Good agreement between the experimental and calculated 1H and 13C NMR chemical shifts of compounds were obtained. The 13C NMR correlation value (R2) of molecule 7a confirms this agreement. Thermodynamic analysis indicated that these compounds are stable in gas phase. According to the NBO analysis, the greatest resonance energy (E(2)) was obtained for LP(1)N7 that participates as electron donor and the anti-bonding BD*(2)(C8-O10) orbital as acceptor. This result shows that charge is transferred from N10 to C11-O20 (N10 → C11-O20). The FMO analysis suggests that charge transfer is taking place within the molecule and the HOMO is focused mainly on isatin ring and phenyl ring whereas the LUMO resides on the isatin ring. The HOMO-LUMO measured energy gap (Eg) for 7a was 10.09 eV.
Supplementary material
The IR, MS and NMR spectra of these compounds are provided as supplementary material. The Cartesian coordinates of the optimized structures of these compounds are also provided with that.
Acknowledgements
We thank from research council of Young Researchers and Elite Club of Islamic Azad University, Gorgan Branch, Iran, for financial support.
References
1 M.A. De La Cruz, H. Shabany and D.C. Spilling, Phosphorus, Sulfur Silicon Relat. Elem., 1999, 146, 181-184. [ Links ]
2 H.D. Durst, D.K. Rohrbaugh and S. Munavalli, Phosphorus, Sulfur Silicon Relat. Elem., 2009,184, 2902-2909. [ Links ]
3 G. Cera, P. Crispino, M. Magda Monari and M. Bandini, Chem. Commun., 2011,47, 7803-7805. [ Links ]
4 I. Yavari, N. Hazeri, M.T. Maghsoodlou and S.J. Souri, Mol. Catal. A -Chem., 2007, 264, 313-317. [ Links ]
5 A. Alizadeh, S. Rostamnia and L.G. Zhu, Tetrahedron, 2006, 62,56415644. [ Links ]
6 A. Shaabani, E. Soleimani and A. Maleki, Tetrahedron Lett., 2006, 47, 3031-3034. [ Links ]
7 A. Shaabani, M.B. Teimouri and H.R. Bijanzadeh, Tetrahedron Lett., 2002,43, 9151-9154. [ Links ]
8 A. Shaabani, I. Yavari, M.B. Teimouri, A. Bazgir and H.R. Bijanzadeh, Tetrahedron, 2001, 57, 1375-1378. [ Links ]
9 S.M. Habibi-Khorassani, M.T. Maghsoodlou, N. Hazeri, K. Bagher-pour, M. Rostamizadeh and H. Najafi, Phosphorus, Sulfur Silicon Relat. Elem., 2010,186, 1395-1403. [ Links ]
10 (a) A.A. Esmaeili, R. HosseinabadiandM. Razi, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 2267-2273. [ Links ] b) D. Azarifar and D. Sheikh, Synth. Commun., 2013,43, 2517-2526. [ Links ]
11 U. Bora, A. Saikia and R.C. Boruah, Org. Lett., 2003, 5,435-438. [ Links ] b) D. Azarifar, D. Sheikh, Helv. Chim. Acta, 2012, 95, 1217-1225. [ Links ]
12 H.M.R. Hoffman and J. Rabe, Helv. Chim. Acta, 1984, 67, 413-415. [ Links ]
13 F. Ameer, S.E. Drewes, N.D. Emslie, P.T. Kaye and R.L. Mann, J. Chem. Soc. Perkin Trans. 1, 1983, 28, 2293-2295. [ Links ]
14 M. Paira, B. Banerjee, S. Jana, S.K. Mandal and S.C. Roy, Tetrahedron Lett., 2007, 48, 3205-3207. [ Links ]
15 H.M.R. Hoffmann and J. Rabe, J. Org. Chem., 1985, 50, 3849-3859. [ Links ]
16 E.K. Scott, B. Martin, T. Raj and K. Gall, Polymer., 2009,50,5549-5558. [ Links ]
17 L. Lienafa, S. Monge andJ.J. Robin, Eur. Polym.J., 2009,45,1845-1850. [ Links ]
18 D. Baskaran, Prog. Polym. Sci., 2003, 28, 521-581. [ Links ]
19 L. Feng, Z. Zhang, F. Wang, T. Wang and S. Yang, Fuel Processing Technol., 2014,118, 42-48. [ Links ]
20 H. Bakhshi, M.J. Zohuriaan-Mehr, H. Bouhendi and K. Kabiri, Polymer Testing, 2009, 28, 730-736. [ Links ]
21 C.J. Cramer, Essentials of Computational Chemistry: Theories and Models, Wiley, Chichester, 2002. [ Links ]
22 D. Avci and Y.Atalay, Struct. Chem.,2009,20,185-201. [ Links ] b) R.B.Nazarski, J. Phys. Org. Chem., 2009, 22, 834-844. [ Links ]
23 S.M. Shoaei, A.R. Kazemizadeh and A. Ramazani, Chin. J. Struct. Chem., 2011, 30, 568-574. [ Links ]
24 E. Vessally, Russ. J. Phys. Chem. A., 2011, 85, 245-247. [ Links ]
25 H. Hopfl, B. Gomez and R. Martinez-Palou, J. Mex. Chem. Soc., 2005, 49, 307-311. [ Links ]
26 G.S. Suresh Kumar, A. Antony Muthu Prabu, S. Jegan Jenniefer, N. Bhuvanesh, P. Thomas Muthiah and S. Kumaresan, J. Mol. Struct., 2013,1047, 109-120. [ Links ]
27 J. Casanovas, A.M. Namba, R. da Silva and C. Aleman, Bioorg. Chem., 2005, 33, 484-492. [ Links ]
28 H. Khanmohammadi and M. Erfantalab, Spectrochim. Acta A, 2010,75, 127-133. [ Links ]
29 H. Khanmohammadi, H. Keypour, M. SalehiFard and M.H. Abnosi, J. Incl. Phenom. Macro., 2009, 63, 97-108. [ Links ]
30 I.YavariandM.TMaghsoodlou, Tetrahedron Lett.,1998,30,4579-4580. [ Links ]
31 A. Ramazani, P. Pakravan, M. Bandpey, N. Noshiranzadeh and A. Souldozi, Phosphorus, Sulfur, and Silicon, 2007,182,1633-1640. [ Links ]
32 A. Ramazani, A. Safari and N. Noshiranzadeh, Int. J. Sci. Technol., 2009,16, 7-11. [ Links ]
33 R.P Gangadharana and S.S. Krishnanb, Act. Phys. Polon. A, 2014,125, 18-22. [ Links ]
34 B.D. Joshi, P. Tandon and S. Jain, Himalayan Phys., 2012, 3,44-49. [ Links ]
35 A.E. Reed, L.A. Curtiss and F. Weinhold, Chem. Rev., 1988,88,899-926. [ Links ]
36 P. Politzer and D.G. Truhlar, Chemical Applications of Atomic and Molecular Electrostatic Potentials, Plenum Press, New York, 1981. [ Links ]
37 PK. Chattaraj, U. Sarkar and D.R. Roy, Chem. Rev., 2006,106,2065-2091. [ Links ]
38 M.J.Frisch, G.W Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery, R.E. Stratmann, J.C. Burant, S. Dapprich, J.M. Millam, A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi, V. Barone, M Cossi, R, Cammi, B Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G.A. Petersson, P.Y. Ayala, Q, Cui, K. Morokuma, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B. Raghavachari, J. Cioslowski, J.V. Ortiz, A.G. Baboul, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T. Keith, M.A. Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, J.L. Andres, C. Gonzalez, M.H. Gordon, E.S. Replogle and J.A. Pople, Gaussian 98 Revision A.7, Gaussian, Inc., Pittsburgh PA, 1998. [ Links ]
39 a) A.D. Becke. J. Chem. Phys., 1993, 98, 5648-5652. [ Links ] b) C. Lee. W. Yang and R.G. Parr, Phys. Rev. B, 1998, 37, 785-789. [ Links ]
40 a) M. Monajjemi, M. Sheikhi, M. Mahmodi Hashemi, F. Molaamin and R. Zhiani. Inter. J. Phys. Sci., 2012, 7, 2010-2031. [ Links ] b) S.A. Seyed Katouli, M. Sheikhi, D. Sheikh and S.F. Tayyari, Orient J. Chem., 2013, 29, 1121-1128. [ Links ]
41 M. Monajjemi, S. Afsharnezhad, M.R. Jaafari, T. Abdolahi, A. Niko-sade and H. Monajjemi. Phys. Chem. Liquids, 2011,49, 318-336. [ Links ]
42 a) M. Monajjemi, L. Mahdavian, F. Mollaamin and B. Honarparvar. Fullerenes, Nanotubes. Carbon Nano-structures, 2010, 18, 45-55. [ Links ] b) L. Shiri, D. Sheikh, A.R. Faraji, M. Sheikhi and S.A. Seyed Katouli, Lett. Org. Chem., 2014, 11, 18-28. [ Links ]
43 A.R. Soltani, M.T. Baei, M. Mirarab, M. Sheikhi and E.Tazikeh Lemeski, J. Phys. Chem. Solids., 2014, 75, 1099-1105. [ Links ]
44 A. Frisch, A.B. Nielson and A.J. Holder, GAUSSVIEW User Manual, Gaussian Inc., Pittsburgh, PA, 2000. [ Links ]
Received 13 July 2014
Revised 23 September 2014
Accepted 24 September 2014.
* To whom correspondence should be addressed. E-mail: m.sheikhi2@gmail.com


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}