










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban Jan. 2014
RESEARCH ARTICLE
Determination of Carboxylic Acids and Water-soluble Inorganic Ions by Ion Chromatography in Atmospheric Aerosols from Tanzania
Stelyus L. MkomaI, *; Gisele O. da RochaII; Jailson B. de AndradeII
IDepartment of Physical Sciences, Faculty of Science, Sokoine University of Agriculture, P.O. Box 3038, Chuo Kikuu, Morogoro, Tanzania
IIInstituto de Química, Universidade Federal da Bahia (UFBA), Campus de Ondina, 40170-290, Salvador-BA, Brazil
ABSTRACT
Atmospheric aerosol samples of PM2,5 and PM10 were collected in April-May 2011 from a rural site in Tanzania and analyzed for water-soluble inorganic ions and low molecular weight carboxylic acids using ion chromatography. PM2,5 and PM10 low-volume samplers with quartz fibre filters were deployed and aerosol collections on a 24 h basis were made for each sampler. All samples were analyzed for carboxylates and inorganic ions by using ion chromatography. The results showed that mean mass concentration of PM2,5 and PM10 were 13 ± 3.5 µg m-3 and 16 ± 2.3 µg m-3, respectively. Mean concentrations of the total carboxylates were 23.7 ± 6.5 ng m-3 in PM2.5 and 36.4 ± 12 ng m-3 in PM10 whereas total water-soluble inorganic ions were 448 ± 88 ng m-3 and 646 ± 214 ng m-3, respectively. Oxalate and malonate in PM25 and acetate in PM10 were most abundant carboxylates accounting for 64 % and 62 % of total acids, respectively. Mg2+ was most important cation in PM25 and PM10 accounting for 44 % and 24 % of total water-soluble ions, respectively, whereas SO42- was the main anionic component accounting for 23 % of total ions species in PM25 and 37 % in PM10. Using source indicators, we found photochemical activities, biogenic and biomass burning could possibly be important source types for aerosols in Tanzania.
Keywords: PM2.5, PM10, aerosol particles, organic acids, sources, Tanzania.
1. Introduction
Carboxylic acids together with water-soluble inorganic ions are an important group of water-soluble organic compounds in atmospheric aerosols.1,2 They are highlighted because they account for a substantial portion of atmospheric aerosols, and potentially control chemical and physical properties of the particles. Consequently, they may have direct and indirect effects on the earth's radiation balance by scattering incoming solar radiation, which counteracts global warming.3 More attention has been paid to carboxylic acids due to their potential to modify the hygroscopic properties of atmospheric particles, including cloud condensation nuclei activity and hence to change global radiation balance.4,5 Major water-soluble inorganic ions are associated with atmospheric visibility degradation, adverse human health effects, and acidity of precipitation.3,6
Chemical composition of PM2.5 and even that of PM10 aerosols is important to gain insights into sources and of their toxicity and to evaluate effectiveness of abatement strategies for relevant emission sectors. Among the organic acids, low molecular weight carboxylic acids such as acetic, oxalic and malonic are generally most abundant in the atmospheric aerosols. Carboxylic acids in variable concentrations have been reported in various environments including rural and urban atmosphere7,8,9,10 and have different source origin, including biomass burning, fossil fuel combustion,11,12 sea spray, traffic and industrial emissions and photochemical oxidation of precursors from anthropogenic and biogenic origin.13,14 Other sources for carboxylic acids in the marine atmosphere include in-cloud and heterogeneous formations.15
In Africa aerosols measurements especially of organic components have not been carried out extensively. Therefore, a full scenario of air quality is far from being revealed because some pollutants including carboxylic acids have not been measured. Here we report for the first time in Tanzania, composition of low molecular weight carboxylic acids in aerosol samples collected from a rural background atmosphere in Morogoro. An insight of characteristics of water-soluble inorganic ions is also discussed.
2. Experimental
2.1. Aerosol Sampling Site
Aerosol samples were collected at a rural site in Morogoro (300 000 inhabitants) for two weeks in April-May 2011. This site (Fig. 1) is located at about 200 km west of the Indian Ocean and the city of Dar es Salaam, a business capital in Tanzania. The samples were collected at Solomon Mahlangu Campus of Sokoine University of Agriculture (06°47'41"S, 37°37'44"E, altitude 504 m a.s.l.). This site is located about 6 km from Morogoro central area and major road systems and possible aerosol sources include biomass burning, agriculture, livestock and soil dust. Tropical savanna is the most important land cover in large part of the sampling site.
2.2. Aerosol Collection
A 'Gent' PM25 and PM10 filter holders were used in parallel to collect aerosol particles using quartz fibre filters (Whatman QM-A) which were pre-fired at 550 °C for 24 h before use. Samplers operated at a flow rate of 17 L min-1 and sampling was carried out approximately at 24 h intervals and exchange of filters was done at 07:30. A total of 11 sets of filed samples and two blanks were collected for each sampler. After sampling the exposed filters were folded in half face to face, placed in polyethylene plastic bags and kept frozen at -20 °C during storage and transported cool to the Laboratory of Research and Development in Chemistry at the Institute of Chemistry, Federal University of Bahia. The samples were stored in a freezer at -20 °C prior to analysis. All the procedures were strictly quality-controlled to avoid any possible contamination of the samples.
During the sampling period meteorological data were collected at site. The daily winds were predominantly south-easterly with an average speed of 6.8 m s-1. Average temperature was 26.8 °C and average relative humidity was 73 %. The recorded maximum temperature and relative humidity were 29.8 °C and 79.5 %, while minimum were 23.7 °C and 63.5 %, respectively. During the two-week campaign, there were only 5 days with weak rain (19.9 mm).
2.3. Aerosol Analyses
For particulate mass measurements, the filter samples were weighed before and after sampling with an analytical microbalance balance Mettler Toledo MX5 (reading precision 1 μg). Before weighing, the filters were conditioned in a chamber equipped with hydro-thermometer clock at a temperature of 20°C and relative humidity of 40 % for 48 h and the weighings were done under these conditions.
For determination of carboxylic acids and water-soluble ions one-half of 12.88 cm2 portions punched from of each PTFE filter was extracted using 5 mL Milli-Q ultrapure water (resistivity > 18.2 MQcm, Barnstead International, USA) in a shaker tube Mode lAT56 (Fanem Ltd, Säo Paulo, Brazil) for 5 min, followed by filtering through a polytetrafluoroethylene (PTFE) filter (0.45 pore size, Sartorius Stedim, Germany). The concentrations of aqueous extracts were analyzed by Dionex ion chromatography ICS 1100 for carboxylates and anions and ICS 2100 for cations. An analytical column AS16 (3 x 50 mm) with AG16 guard column (3 x 50 mm) and CSRS-3001 (2 mm) suppressor in ion-exchange mode was used to determine carboxylates (monocarboxylates: formate and acetate; dicarboxylates: oxalate, malonate, succinate, and maleate; ketocarboxylate: pyruvate) and water-soluble anions (chloride Cl-, nitrate NO3-and sulphate SO42-). The eluent gradient programme was sweeping from 6.0 to 8.0 mmol L-1 KOH (potassium hydroxide) in 35 min under flow rate of 0.38 μL min-1, except for acetic acid which was determined in another run, reducing injection time to avoid overlap of peaks.
For determination ofwater-soluble cations (ammonium NH4+, sodium Na+, potassium K+, magnesium Mg2+ and calcium Ca2+) an analytical column CS16 and Guard column CG16 (both 3 x 50 mm) and CSRS-I (2 mm) suppressor in a chemical mode were used. An eluent of 17.5 mmol L-1 H2SO4 (sulphuric acid) was used at flow rate of 0.35 μL min-1. The injection volume was 25 μL for all detection. Peak identification was confirmed based on a match of ion chromatograph retention times and standard samples. Limit of detection determined as mean equal to three times standard deviation of the field blank value corresponded to a range of 0.008 to 0.017 ng L-1 for carboxylates, 0.008 to 0.023 ng L-1 for anions and 0.021 to 0.083 ng L-1 for cations. Limits of quantification were between 0.026 and 0.058 ng L-1 for carboxylates, 0.028 and 0.078 ng L-1 for anions and 0.063 and 0.252 ng L-1 for cations.
3. Results and Discussion
3.1. Concentrations of PM Mass
Mean PM mass concentrations and associated standard deviations and ranges as derived from the two low-volume samplers are shown in Table 1. The results showed that mean mass concentration of PM2.5 and PM10 aerosols during the campaign were 13 ± 3.5 μg m-3 and 16 ± 2.3 μg m-3, respectively. The percentages of PM25 mass in PMW size fraction (Fig. 2) found to range from 44-99 % with a mean of 83 ± 29 %. These results indicate that most of PM mass was in PM2.5 size fraction. High PM25/PM10 ratios for PM mass indicate that there is small contribution from soil dust, which is known to be mostly associated with PM10 aerosols. Currently in Tanzania, the ambient air quality standard limit values for inhalable particulate matter are 60 to 90 μg m-3 for PM10.16 The mean concentrations for PM10 mass at our site in Morogoro were below these average limit values. In addition, the current data sets were in line with levels reported in our previous studies.17,18,19 Nevertheless, when compared PM mass data from our rural site in Tanzania are in line with few available other data sets for rural sites in southern Africa.20 They are also comparable to or lower than other sites in Europe and Asia.21,22,23,24
3.2. Concentrations of Carboxylates Ions
Table 1 presents mean total concentrations and range of carboxylates (TCAs) which were 23.7 ± 6.5 ng m-3 (range: 13.3-36.5ngm-3)inPM25and36.4 ± 12ngm-3 (range: 10.7-58.2 ngm-3) in PM10 aerosols. Oxalate and malonate were most abundant carboxylates in PM25 accounting for 32.5 % and 31.85 % of total carboxylates, respectively, whereas in PM10 acetate was most abundant accounted for 62.5 % of total carboxylates followed by oxalate which accounted for 32.6 % of total carboxylates. Other studies have also reported oxalates to be most abundant carboxylate in aerosol samples.15,25 Pyruvate was also found in substantial amount and formate the least abundant counting on average 3 % of total carboxylates in each of the aerosol fractions. Succinate and malonate were below detection limit in PM25 and PM10 aerosols, respectively. The total carboxylates accounted for 0.18 % to total PM25 mass and 0.22 % to PM10 mass. In comparison with other studies, the mean concentrations of the measured carboxylates in Tanzania were lower than those reported in other urban and rural sites around the world.7,9,26,27
3.3. Water-soluble Inorganic Ions
Chemical characteristics of water-soluble inorganic ions and their relative abundances in PM25 and PM10 aerosols are also shown in Table 1. In both aerosol fractions, water-soluble Mg2+ was the most important cation and SO42- the main anionic species. On average Mg2+ accounted for 44.4 % of total water-soluble ions in PM25 and 24.7 % in PM10 whereas SO42-accounted for 22.8 % and 35.2 % of total ions in PM25 and PM10, respectively. High levels of crustal element Mg2+ together with Ca2+ are essentially attributable to soil/mineral dust dispersal. As to reasonable NH4+ levels (8 % of total ions) in PM2.5, this may be due to presence of ammonia gas from biomass burning especially during smoldering combustion28 and from agricultural activities in particular cattle raising.29,30 Water-soluble K+, a good indicator for biomass burning, was second most abundant cation in PM2.5 accounted for 10.6 % of total water-soluble ions.
For SO42 the higher levels could be attributed to its efficient formation by in-cloud processing of SO228 and from secondary formation processes.31 As to low NO3 levels, this is likely due to the fact that the site is rural with little or no traffic and undoubtedly there are less anthropogenic emissions of precursor gas NOx. Also as to low concentrations of Na+ which is mainly derived from sea-salt, this is presumably due to long distance (about 200 km) from the Indian Ocean to our sampling site. The observed levels for water-soluble ions are comparable with those reported in our previous work in Morogoro.1719 It appears that the levels of SO42-, NO3-, and NH4+ in PM10 fractions are substantially lower in Tanzania than at European rural sites32 and Asia.33,34
To determine the impact of marine sources on chemical composition of aerosol particles in PM2.5 and PM10 fractions, sea-salt ratios were calculated for each inorganic ion using Na as a reference element, assuming all Na to be of marine origin. The ratios for Cl/Na+, SO42/Na+, K+/Na+, Mg2+/Na+ and Ca2+/Na+ in PM25 were 0.15 (0.25), 6.48 (1.81), 2.95 (0.04), 12.97 (0.04), 2.44 (0.12), respectively. The corresponding values in PM10 were 0.10 (0.25), 2.51 (1.81), 0.43 (0.04), 1.68 (0.04), 0.75 (0.12), respectively. Values in brackets represent mean ratios for each ion in sea-water.35 Larger ratios of ions indicate incorporation of non-marine constituents in aerosols. As to low mean Cl-/Na+ ratios than sea-water ratio indicates that a minor fraction of Na+ may be contributed from other sources such as mineral dust. But also low ratio could be due to modifications of sea-salt fraction by non-marine constituents. Chloride loss may be explained by heterogeneous reaction of airborne sea-salt with acidic gases and aerosol species.36
3.4. PM2.5 to PM10 Ratios
The mean PM2.5 (fine) to PM10 (coarse) percentage ratios and associated standard deviations for PM mass, carboxylates and various water-soluble inorganic ions are shown in Fig. 2. The ratios were calculated on the basis of data for PM2.5 and PM10 samples taken in parallel and then averaged over all samples from the sampling period. The mean fine to coarse ratios for all species with exception of those for acetate, Na+, Ca2+, NO3 and Cl were predominantly associated with fine fraction (for more than 55 %). High fine/coarse ratio for PM mass maybe due to a lower contribution from soil dust, which is known to be mostly associated with coarse particles. For carboxylates, high ratios (even larger than 70 %) are considered to be attributed from secondary organic aerosols (SOA), biomass burning activities and high temperature (average: 26.8 °C during sampling period). Concentrations of oxalate in PM25 showed strong correlations (r2 = 0.70) with those in PM10 aerosols. The slope of linear regression equations (PM25 = 2.48 x PM10) indicated that oxalate was mainly present in fine fraction during sampling period. On the other hand, different size distributions between oxalate and acetate could be related to their different physical characteristics. Acetate in PM2.5 fraction could easily volatilize (more volatile than oxalate) to gas phase, part of which could be absorbed on PM10 particles.
For water-soluble inorganic ions, as expected, sea-salt elements (Na, Cl) and indicator element for crustal matter (Ca) were predominantly (for more than 62 %) associated with PM10 aerosols. NH4+ and non-sea-salt sulphate, nss-SO42- were mainly present in the fine aerosols suggesting that these species originated from high temperature sources and/or gas-to-particle conversion. The nss-SO42- is due to oxidation of SO2, which is predominantly from anthropogenic origin (e.g. biomass burning). A well-known indicator for biomass burning, K was associated with the fine particles (about 100 %) suggesting vegetative emissions and crustal source could be an important source for K+ aerosols at this site with small impact from biomass burning activities.
3.5. Sources Identification of Carboxylates and Inorganic Ions
3.5.1. Correlation Analysis
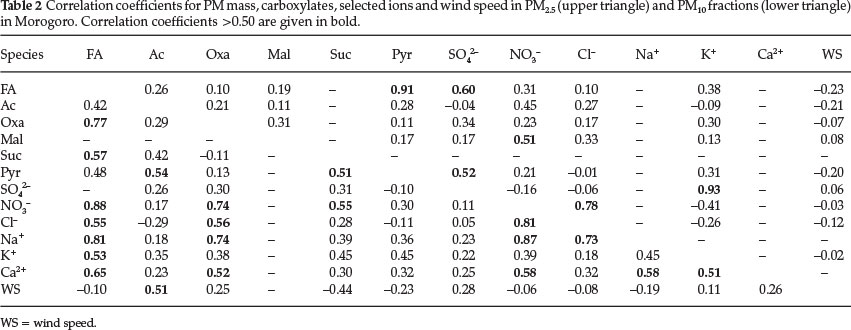
Correlation coefficients of PM mass, carboxylates and source indicators, shown in Table 2 were performed in order to understand their possible sources and formation mechanisms. The selected source indicators include K+ for biomass burning and vegetation emissions, Na+ and Cl- for sea spray or waste burning, and SO42- for secondary formation of different mechanisms. Temperature and wind speed have been used as additional parameters to illustrate the atmospheric behaviours of carboxylic acids. In this study K+ had good correlation with formate (r2 = 0.53) and moderate to poorly correlations with other carboxylates in PM10. This indicates that formate could be originated from biomass and/or waste burning emissions but other carboxylates are considered to have other important sources than biomass burning. It can also be observed from Table 2 that there were possible similar sources for formate and other carboxylates (oxalate, succinate and pyruvate) as verified by good correlation between them in PM2.5 and PM10 aerosols. Pyruvate shows good correlation with acetate (r2 = 0.54) and succinate (r2 = 0.51) in PM10 aerosols. These indicate a feature of photochemical decomposition of succinic acid.37
Sea-salt derived aerosols have been reported to have particles with aerodynamic diameters between 1 and 5 μm.7 In this study, we found Na+ in pronounced amounts in PM10 aerosol and to a slight extent Cl-, suggesting sea spray could be one of the contributing sources of the aerosol components at the site. But even though Na+ correlates well (R2 = 0.73) with Cl-, the calculated Cl- to Na+ mass ratio to sea-salt component in aerosol particles had mean values between 0.15 (PM2.5) and 0.10 (PM10). This suggests that continental contributions were more important than marine contribution since Cl-/Na+ ratio in marine aerosols varies between 1.0 and 1.7.38
Sulphate has been used as reference to investigate major formation routes of carboxylic acids.39 As shown in Table 2, formate and pyruvate showed good correlation with SO42-in PM2.5, suggesting that in-cloud and heterogeneous formations play an important role in the formation of carboxylates. On the other hand, poor correlation of malonic with SO42- in PM2.5 suggests that possibly the acid is volatile at ambient tempera-tures.5 Acetate and oxalate showed poor correlations with SO42-in both aerosol fractions, indicating that they mainly originated from primary emissions sources and/or the atmospheric processes different from those of SO42-. This is contrary to what has been observed in other studies that in-cloud and heterogeneous formations can yield a good correlation between oxalate and SO42-.39
Wind speed poorly correlated with most carboxylates except with acetate (r2 = 0.51) in PM10 aerosols. This indicates that in addition to secondary formation, carboxylates were mainly generated from local sources, while acetate might be related to long-range aerosol transport to the sampling site. It should also be noted that primary emissions are major sources of precursors for most carboxylic acids.9
3.5.2. Concentrations Ratios
The ratio of acetic to formic acid has been used as good indicator of contributions of primary (high ratio) and secondary sources (low ratio) to carboxylic acids.40,41 In this study the acetate/ formate ratios ranged from 0.07-1.07 (mean: 0.24 ± 0.31) in PM25 and 0.03-0.10 (mean: 0.05 ± 0.03) in PM10 aerosols particles. These low ratios indicate that secondary formation was an important contributing source of carboxylates at our site (high mean average temperature (26.8 °C) during the sampling period).
The ratio of oxalic acid to total dicarboxylic acid (for this study oxalic, malonic, succinic acids) can be used to evaluate aging process of organic aerosols,42 because diacid such as oxalic acid can be produced by oxidations of longer-chain dicarboxylic acids.43 In our study oxalate to total dicarboxylates ratios showed low mean values of 0.33 ± 0.08 (range: 0.23-0.49) and 0.33 ± 0.17 (range: 0.15-0.83) in PM25and PM10, respectively, indicating that aerosols emitted from various sources and transported to this site were less and equally aged. Since there relative humidity was high during the campaign (up to 73 % on average), it is supposed that oxalate was also produced in aqueous phase. Aqueous phase chemistry in aerosol and/or cloud droplets is important in production of oxalic acid.15 On the other hand, mean ratio of oxalate to K+ in PM25 was 0.19 ± 0.08 (range: 0.07-0.36), somewhat closer to or higher than the reported range (0.03-0.1) for flaming and smoldering phases in burning plumes.44 This suggests that carboxylates might have originated from biogenic sources and biomass burning emissions.
4. Conclusion
PM2.5 and PM10 aerosols samples were collected from a rural site at Morogoro, Tanzania, and analyzed for low molecular weight carboxylates and water-soluble inorganic ions. Oxalate and malonate were dominant species in PM2.5 while acetate was the most prominent species in PM10 aerosols followed by oxalate. Of the ionic components, SO42-, K+ and Mg2+ in PM2.5 and SO42-, Na+ and Mg2+ in PM10 made greater contributions to total water-soluble inorganic aerosol mass. Various ratios and correlations between carboxylates and ions suggest that primary emissions, secondary formation, and to a small extent sea spray and biomass burning could be the sources for the aerosols at this site. The ratio of acetate to formate was found to be close to the reported value for secondary reactions, indicating dominance of secondary sources. This study suggests that to better understand the Tanzania atmosphere more work is needed to determine longer-chain (high) molecular weight carboxylic acids and related organic compounds and their seasonal variations in urban and rural sites.
Acknowledgements
The authors acknowledge the financial support from Directorate of Research and Postgraduate Studies, Sokoine University of Agriculture (SUA)-Tanzania and Conselho Nacional de Desenvolvimento Científico e Tecnolýgico (CNPq)-Brazil. We thank Mr. Filbert T. Sogomba of the Department of Physical Sciences (SUA) for sample collection and Cibele Cristina de Araújo Soares (UFBA, Brazil) for PM mass measurements.
References
1 M.C. Jacobson, H.C. Hanson, K.J. Noone and R.J. Charlson, Rev. Geophys., 2000, 38(2), 267-294. [ Links ]
2 C. Bourotte, A.-P. Curi-Amarante, M.-C. Forti, A. Luiz, A. Pereira, A.L. Braga and PA. Lotufo, Atmos. Environ., 2007,41(10), 2036-2048. [ Links ]
3 Intergovernmental Panel on Climate Change (IPCC). IPCC fourth assessment report, Contribution of Working Group I, Cambridge University Press, London, 2007 p. 996. [ Links ]
4 V-M. Kerminen, J. Geophys. Res., 2001,106(D15), 17321-17333. [ Links ]
5 C. Peng, M.N. Chan and C.K. Chan, Environ. Sci. Technol., 2001,35(22), 4495-4501. [ Links ]
6 D. Dockery and A. Pope, in Particles in Our Air: Concentration and Health Effects ,(J.D. Spengler and R. Wilson, eds.), Harvard University Press, Cambridge, MA, 1996, pp. 123-147. [ Links ]
7 V-M. Kerminen, C. Ojanen, T. Pakkanen, R. Hillamo, M. Aurela and J. Merilaien, J. Aerosol Sci., 2000, 31(3), 349-362. [ Links ]
8 A. Limbeck, H. Puxbaum, L. Otter and M.C. Scholes, Atmos. Environ., 2001, 35(10), 1853-1862. [ Links ]
9 K. Kawamuraand O. Yasui, Atmos. Environ., 2005,39(10), 1945-1960. [ Links ]
10 J.F. Nicolas, N. Galindo, E. Yubero, C. Pastor, R. Esclapez and J. Crespo, Water Air Soil Poll., 2009, 201(1-4), 149-159. [ Links ]
11 K. Kawamura and I.R. Kaplan, Environ. Sci. Technol., 1987,21,105-110. [ Links ]
12 M. Narukawa, K. Kawamura, N. Takeuchi and T. Nakajima, Geophys. Res. Letters, 1999, 26(20), 3101-3104. [ Links ]
13 K.F. Ho, S.C. Lee, J.J. Cao, K. Kawamura, T. Watanabe, Y Cheng and J.C. Chow, Atmos. Environ., 2006, 40, 3030-3040. [ Links ]
14 S.G. Aggarwal and K. Kawamura, J. Geophys. Res., 2008,113(D14301) DOI: 10.1029/2007JD009365. [ Links ]
15 P. Warneck, Atmos. Environ., 2003, 37(17), 2423-2427. [ Links ]
16 Tanzania Bureau of Standards, TBS. National Environmental Standards Compendium EMDC 6(1733), p. 74, TZS 845: 2006 Air Quality Specification, 2006. [ Links ]
17 S.L. Mkoma, X. Chi, W. Maenhaut, W. Wang and N. Raes, Atmos. Environ., 2009, 43(3), 631-639. [ Links ]
18 S.L. Mkoma, W. Maenhaut, X. Chi, W. Wang and N. Raes, X-Ray Spectrom., 2009, 38(4), 293-300. [ Links ]
19 S.L. Mkoma, W. Wang, W. Maenhaut and C.T. Tungaraza, Ethiopian J. Environ. Studies Manage., 2010, 3(2), 27-38. [ Links ]
20 D. Nyanganyura, W. Maenhaut, M. Mathuthu, A. Makarau and F.X. Meixner, Atmos. Environ., 2007, 41(12), 2644-2659. [ Links ]
21 R. Van Dingenen, et al., Atmos. Environ., 2004, 38(16), 2561-2577. [ Links ]
22 J. Gu, Z. Bai, A. Liu, L. Wu, Y. Xie, W. Li, H. Dong and X. Zhang, Aerosol Air Qual. Res., 2010,10(2), 167-176. [ Links ]
23 W. Maenhaut, S. Nava, F. Lucarelli, W. Wang, X. Chi and M. Kulmala, X-Ray Spectrom., 2011,40(3), 168-171. [ Links ]
24 K. Ram and M.M. Sarin, Atmos. Environ., 2011, 45(2), 460-468. [ Links ]
25 M. Mochida, A. Kawabata, K. Kawamura, H. Hatsushika and K. Yamazaki, J. Geophys. Res., 2003,108(D6), 4193-4203. [ Links ]
26 S.R. Souza, P.C. Vasconcellos and L.R.F. Carvalho, Atmos. Environ., 1999, 33(16), 2563-2574. [ Links ]
27 X. Yao, M. Fang, C.K. Chan and M. Hu, Atmos. Environ., 2003, 37(21), 3001-3007. [ Links ]
28 M.O. Andreae and P. Merlet, Global Biogeochem. Cycles, 2001, 15(4), 955-966. [ Links ]
29 D.G. Streets, K.F. Yarber, J.-H. Woo and G.R. Carmichael, Global Biogeochem. Cycles, 2003, 17(4), 1099-1118. [ Links ]
30 E.A. Stone, J.J. Schauer, B.B. Pradhan, P.M. Dangol, G. Habib, C. Venkataraman and V. Ramanathan, J. Geophys. Res., 2010, 115(D06301), DOI: 10.1029/2009JD011881. [ Links ]
31 A.G. Allen, A.A. Cardoso and G.O. da Rocha, Atmos. Environ., 2004, 38(30), 5025-5038. [ Links ]
32 J.-P. Putaud, et al., Atmos. Environ., 2004, 38(16), 2579-2595. [ Links ]
33 S.G. Aggarwal and K. Kawamura, Atmos. Environ., 2009, 43(16), 2532-2540. [ Links ]
34 C.M. Pavuluri, K. Kawamura, S.G. Aggarwal and T. Swaminathan, Atmos. Chem. Phys., 2011,11(15), 8215-8230. [ Links ]
35 P.G. Brewer, in Chemical Oeanography, (J.P. Riley & G. Skirrow, eds.), Academic Press, London, UK, 1975, pp. 415-496. [ Links ]
36 F.J. Millero. Chemical Oceanography, 3rd edn, Taylor and Francis, CRC Press, Boca Raton, FL, 2006. [ Links ]
37 X. Yao, M. Fang and C.K. Chan, Atmos. Environ., 2002, 36(13), 2099-2107. [ Links ]
38 R. Chesselet, M. Morelli and P. Buat-Menard, in The Changing Chemistry of the Oceans, (D. Dyrssen and D. Jagner, eds.), Proceedings of Nobel Symposium 20, Wiley-Interscience, New York, 1972, p. 92. [ Links ]
39 J. Yu, X. Huang, J. Xu and M. Hu, Environ. Sci. Technol., 2005, 39(1), 128-133. [ Links ]
40 R.W. Talbot, M.O. Andreae, H. Berresheim, D.J. Jacob and K.M. Beecher, J. Geophys. Res., 1990, 95(D10), 16799-16811. [ Links ]
41 D. Grosjean, Atmos. Environ., 1992, 26A(18), 3279-3289. [ Links ]
42 K. Kawamura and F. Sakaguchi, J. Geophys. Res., 1999, 104(D3), 3501-3509. [ Links ]
43 K. Kawamura, H. Kasukabe and L. Barrie, Atmos. Environ., 1996, 30(10-11), 1709-1722. [ Links ]
44 M.A. Yamasoe, P. Artaxo, A.H. Miguel and A.G. Allen, Atmos. Environ., 2000, 34(10), 1641-1653. [ Links ]
Received 26 November 2012
Revised 6 April 2014
Accepted 25 April 2014
* To whom correspondence should be addressed. E-mail: stelyusm@gmail.com


{kind=link}
{kind=link}
{kind=link}
{kind=link}