Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Chemistry
versión On-line ISSN 1996-840X
versión impresa ISSN 0379-4350
S.Afr.j.chem. (Online) vol.67 Durban ene. 2014
RESEARCH ARTICLE
Synthesis of Novel Piperazine-linked Anthranilic Acids as Potential Small Molecule Kinase Inhibitors
Santanu ChakravortyI; Hanna F. KleinI; Luke E. HodsonII; Matthias RabillierIII; Zhizhou FangIV; Andre RichtersIV; Stephen C. PellyII; Daniel RauhIII, IV, *; Willem A.L. van OtterloI, II, *
IMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO WITS, Johannesburg, 2050, South Africa
IIDepartment of Chemistry and Polymer Science, Stellenbosch University, Private Bag X1, Matieland, Stellenbosch, 7602, South Africa
IIIChemical Genomics Centre of the Max-Planck-Society, Otto-Hahn-Strasse 15, 44137 Dortmund, Germany
IVDepartment of Chemistry and Chemical Biology, Technical University of Dortmund, Otto-Hahn-Strasse 6, 44227 Dortmund, Germany
ABSTRACT
Substituted anthranilic acid and piperazines were used as building blocks to prepare two libraries of compounds, with the aim being that they would exhibit biochemical activity as small molecule kinase inhibitors. The synthesized anthranilamide-piperazine compounds were subsequently tested against a panel of kinases including EGFR, Abl, Akt and Aurora B.
Keywords: Small molecule kinase inhibitors, anthranilic acid, piperazines, EGFR.
1. Introduction
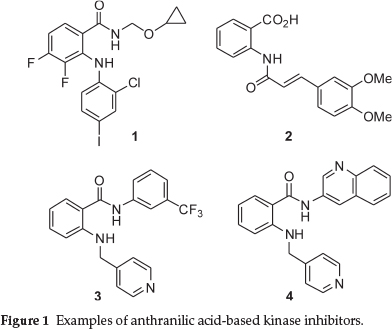
Anthranilic acids and their derivatives represent a structural motif that has seen much application in medicinal chemistry. In the field of oncology, a wide variety of substituted anthranilic acids have been tested and found to have cytotoxic activity against cancerous cells.1-5 In particular, it has been noted that this scaffold is a privileged structure when dealing with kinase inhibition.6,7 Examples include PD184352 1 (aka CI-1040) from Parke-Davis - a MEK1 and MEK2 inhibitor, tranilast 28 and two anthranilic acid amides, AAL993 39 and 4, prepared during a cooperation between Novartis Pharma and Schering AG (Fig. 1). The last three compounds were all found to inhibit vascular endothelial growth factor (VEGF) receptor tyrosine kinase, an important therapeutic target in the field of oncology.10
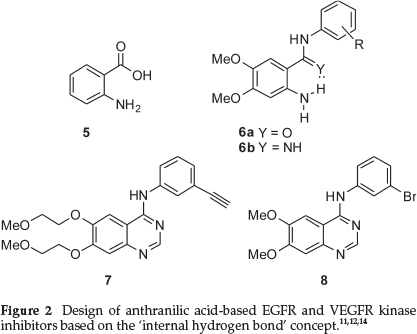
In a 2004 set of papers, anthranilic acid 5 was identified as a possible scaffold for the inhibition of the tyrosine kinases Src and epidermal growth factor receptor (EGFR), resulting in the development of a potent set of benzamides and benzamidines (see for example structures 6 in Fig. 2).11,12 EGFR is a transmembrane protein classified as a 'receptor tyrosine kinase'. This protein is typically activated by the extracelluler binding of a number of ligands, including epidermal growth factor, and has been implicated in a number of important cellular processes. Importantly, its abnormal functioning has also been implicated in numerous human malignancies.13 The same authors extended the study of this class of compounds to demonstrate that vascular endothe-lial growth factor receptors (VEGFRs) were also selectively inhibited by further inhibitors based on the same anthranilic acid scaffold.14 Importantly, the authors of this work proposed that molecules like 6a and 6b most likely possessed a 'slightly different' binding conformation when compared to erlotinib (Tarceva™) 7, although this was not confirmed by an X-ray study.12 Structures 6a and 6b (Fig. 2) have been drawn to show how the presence of an intramolecular hydrogen bond preorganizes 6 to mimic the quinazoline-portion of known EGFR inhibitors such as PD153035 8 and erlotinib (Tarceva™) 7, although it should be realized that the anilineNH2 is unlikely to act as an intermolecular hydrogen bond acceptor as is often proposed for the comparative quinazoline kinase inhibitors. It should be noted that other researchers have also designed potent kinase inhibitors on this particular basis,15,16 and that this topic has become an important concept in medicinal chemistry.17
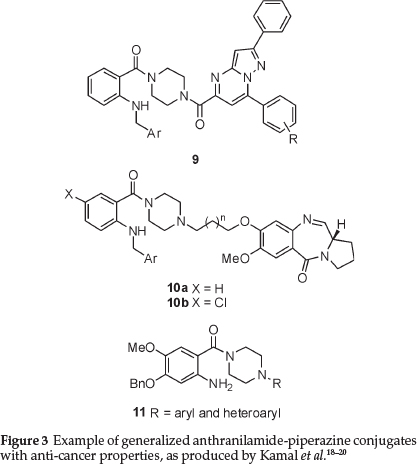
In recent years, the piperazine moiety has seen increasing use as a structural component of compounds with relevance in oncology. As an example, and of particular relevance to this paper, Kamal, Pal-Bhadra and co-workers have recently found that the use of anthranilamide-piperazine conjugates, such as 9 and 10, have demonstrated interesting activity in terms of apoptosis induction in cancer cells (Fig. 3).18,19 These same researchers patented a series of simpler anthranilic acid derivatives with the generic structure 11, as 'potential anticancer agents'.20
Over the past years, our respective research groups have been interested in the design, synthesis and evaluation of small molecules for the selective inhibition of topical kinases,21,22 as well as the associated biochemical evaluations.23-25 The literature concerning the use of intramolecular hydrogen bonds, as well as the innovation of using the piperazine fragment as part of the scaffold,26,27 prompted us to combine these two structural features, resulting in the synthesis of two compound libraries. In the present work, twelve compounds based on the anthranilic acid skeleton and incorporating a piperazine within their structure were synthesized following a three-step protocol, to afford potential kinase-inhibiting anilines which were biochemically evaluated. Secondly, a focused library based on the piperazin-1-yl{2-[(pyridin-4-ylmethyl)amino]phenyl}methanone scaffold was also generated.
2. Synthesis
For the first set of potential inhibitors, 4,5-dimethoxy-2-nitro-benzoic acid 12 was converted into the corresponding acid chloride derivative, after which treatment with a number of substituted anilines and piperazines afforded the nitroamides 13a-l in good to excellent yields over two steps (see Scheme 1 and Table 1 for details). Subsequent reduction of the nitro functional group led to the desired anthranilic acid derivatives 14a-l. These compounds were all thoroughly characterized by spectroscopic techniques which included 1H and 13C NMR, as well as high resolution mass spectroscopy (HRMS).
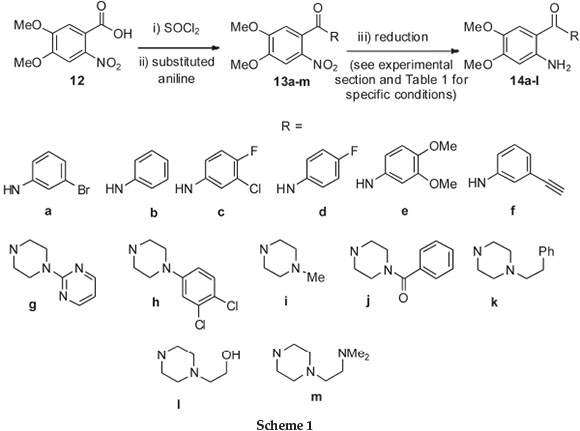
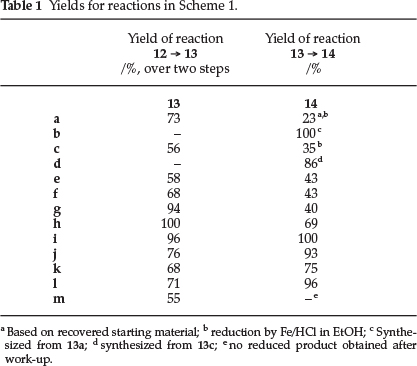
At this point, it should also be stressed that compounds 14a and 14c were synthesized as potential reference compounds, as they were studied in the key Nakamura study mentioned earlier.12 It should also be mentioned here that the palladium-mediated reduction of substrates 13a and 13c did not result in the expected anilines 14a and 14c, but gave compounds 14b and 14d instead, in which the bromine and chlorine atoms had been reductively removed respectively. Application of an alternative reduction method making use of iron in an acidic media, did however afford the desired compounds 14a and 14c, albeit in less than satisfactory yields.
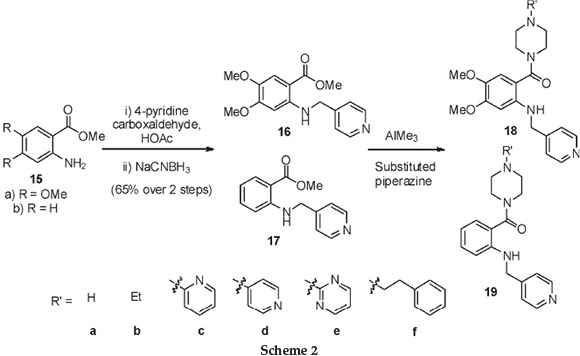
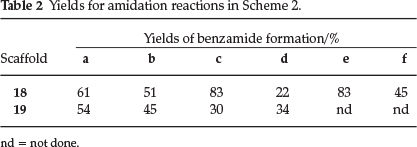
Furthermore, a second set of anthranilic acid analogues, this time containing pyridyl appendages, inspired by compounds 3 and 4, was also synthesized. These compounds were produced by an initial reductive amination between the anthranilic ester 15 and 4-pyridine carboxaldehyde to afford the alkylated anilino scaffolds 16 and 17, respectively (Scheme 2 and Table 2). Furthermore, conversion of the ester functional groups into the corresponding amides, facilitated by the Lewis acid trimethyl aluminium, then afforded the desired anthranilic acid derivatives 18 and 19. These compounds thus featured a pyridine moiety and were decorated with a range of substituted piperazines, as shown in Scheme 2 and Table 2.
3. Biochemical Testing
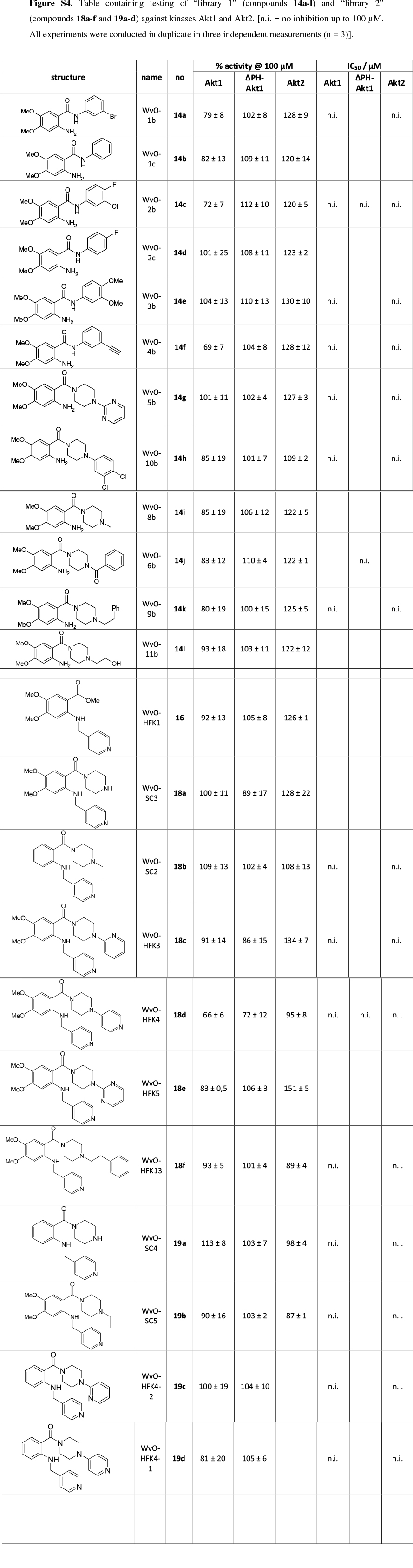
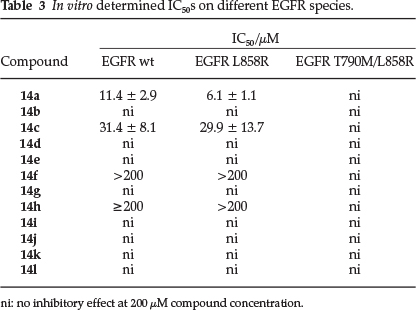
As an initial screening, 12 compounds from 'Library 1' (14a-l) were evaluated for their ability to inhibit the kinase EGFR, which is commonly involved in carcinogenicity.28 Mutations in EGFR account for 10-17 % of all non-small cell lung cancers (NSCLCs), with L858R and short in-frame deletions of exon 19 representing the most common activating mutations.29 In contrast to other EGFR mutations involved in cancer, L858R does not cause resistance to TKI-based therapy, but instead improves the response to first line tyrosine kinase inhibitors such as gefitinib. Secondary mutations providing resistance to small molecule therapy often appear later, as in the case of T790M.30 EGFR is a prime target in cancer therapy, which is emphasized by the large number of small molecules that have been developed to target this receptor tyrosine kinase.31 Furthermore, it is closely related to VEGFR on which the anthranilic scaffold originally was tested. For these reasons, EGFR wt as well as its L858R and T790M/L858R mutated species were used for testing in a biochemical assay setup.22 The results displayed in Table 3 were unfortunately disappointing, with only compounds 14a (~10µM) and 14c (~30µM),from the Nakamura study,12 having any interesting activity on wt and L858R mutated EGFR. The synthesized novel compounds 14f and 14h also displayed slight inhibitory activity >200 µM) in this assay. EGFR harboring both activating (L858R) and gatekeeper (T790M) mutation was not affected at all, up to compound concentrations of 200 µM.
In a further screen, it was decided to test the inhibition of the compounds from both libraries/sets against the Abl T315I kinase at a concentration of 10 /M. This particular kinase has been of interest due to the lack of inhibitors available to target this specific resistance mutation in chronic myelogenous leukemia (CML). Ponatinib (Iclusig), which was recently approved by the FDA, represents the only therapeutic to effectively target Abl T315I, but comes along with severe side effects, indicating a rather complex pharmacology.32 The results from this assay were not encouraging, with none of the compounds demonstrating inhibition at this concentration, although the reference compound, staurosporine, displayed the expected inhibitory effect (See supplementary information, Fig. S1).
It was then decided to send two representative compounds from the first set, namely compounds 14a and 14h, which had shown some activity in the EGFR screen, for testing against the Dundee kinase library, which comprises 95 different kinases (see supplementary information, Figs S2 and S3).33 Of interest was that 14a showed good inhibition of AKT2 (aka PKBb) (17 % remaining activity at 10 µM concentration), as well as moderate inhibition of HER4 and Aurora B (43% resp. 51% remaining activity at 10 µM). Compound 14h also inhibited AKT2 (PKBβ) with moderate effect (64 %), as well as Aurora B (61 %) and CAMK1 (60 %). The two libraries, comprising compounds 14a-l, 18a-f and 19a-d, were then evaluated for their ability to inhibit Akt2 (PKBβ) and its close isoform Akt1 (PKBa), with the enzymes at full length (Akt1 and Akt2) or kinase domain only (Akt1). Unfortunately, the results showed that the compounds did not effectively inhibit the kinases either (see supplementary information, S4).34 Finally, as the Dundee screen had also shown 14a and 14h to inhibit Aurora B, both compounds were sent for testing against this particular kinase to Reaction Biology Corp.35 Compounds 14a and 14h were subsequently tested in 10-dose IC50 mode with threefold serial dilution starting at 300 µM and in the presence of 20 µM ATP. Unfortunately, both compounds showed little activity (IC50 >300 µM)36 in comparison to the positive control staurosporine, which had an IC50 of 5 nM.
4. Conclusion
The present paper presents the synthesis of anthranilic acid-derived molecules inspired by known kinase inhibitors such as Tranilast and AAL993. A particularity of our set of molecules is the piperazine fragment included in their core structure. The synthesized anthranilamide-piperazine conjugates were then tested against the following kinases: EGFR (wt, L858R and T790M/L858R), Abl_T315I, Akt1 (PKBα), Akt2 (PKBβ) and Aurora B. These tests indicated that unfortunately none of the compounds inhibited the kinases enough to warrant further extension of these libraries.
Acknowledgements
W.A.L.v.O. thanks the Alexander von Humboldt Foundation for a Georg Forster Research Fellowship for Experienced Researchers and the University of the Witwatersrand for sabbatical leave. This work was also supported by the National Research Foundation (NRF), Pretoria, the University of the Witwatersrand (University and Science Faculty Research Councils) and Stellenbosch University (Faculty and Departmental funding). The SASOL 'Hub and Spoke' initiative is acknowledged for funding, and S.C. thanks the NRF for a postdoctoral fellowship. We also gratefully acknowledge Mr R. Mampa (University of the Witwatersrand) and Dr J. Brand and Ms. E. Malherbe (Stellenbosch University) for the NMR spectroscopy service. Finally, Mr T. van der Merwe (University of the Witwatersrand), Dr A. Dinsmore and Mrs M. Ferreira (University of the Witwatersrand, LRMS), Ms J. Schneider and Ms M. Ismail (Mass Spectroscopy Service, University of Dortmund and Max Planck Institute for Molecular Physiology, Dortmund, Germany) and Mr B. Moolman, Mr F. Hiten and Dr M. Stander (Stellenbosch University) are gratefully acknowledged for providing MS spectroscopy services.
Experimental
General Experimental Information
1H NMR and 13C NMR spectra were recorded on Bruker 300, Bruker DRX 400, Varian Inova 400 or Varian Inova 300 spectrometers at the frequency indicated. Infra-red spectra were recorded on Bruker IFS 25, Bruker Vector 22 or Thermo Nicolet Nexus 470 fourier transform spectrometers. Mass spectra were recorded on a Kratos MS 9/50, VG 70E MS or VG 70 SEQ mass spectrometer or alternatively a Waters API Q-TOF Ultima, GCT Premier or SYNAPT G2 mass spectrometer. Prior to being evaluated for HRMS all compounds were checked by LCMS for a purity of >80 %. Macherey-Nagel kieselgel 60 (particle size 0.063-0.200 mm) was used for conventional silica gel chromatography. All solvents used for reactions and chromatography were distilled prior to use. Reactions were performed under a blanket of inert gas (Ar or N2) unless specified. Melting points are uncorrected.
General Experimental Procedure for the Synthesis of N-phenylbenzamides 13a-m from Benzoic Acid 12
4,5-Dimethoxy-2-nitrobenzoic acid 12 (0.23 g, 1.0 mmol) was dissolved in SOCl2 (1 mL) and the reaction was stirred at 75 °C for 3 h under an Ar atmosphere. The excess SOCl2 was then removed under reduced pressure using a liquid nitrogen trap. CH2Cl2 (5 mL) was then added to the remaining residue, followed by the addition of the substituted aniline (1.1 mmol, 1.1 mol equiv.) and NEt3 (0.41 mL, 3.0 mmol) and the reaction was stirred at RT, under Ar for 18 h. The organic solvent was removed under reduced pressure and EtOAc (20 mL) was added. The organic fraction was washed sequentially with HCl (10 mL, 1 M), H2O (20 mL), aq. NaOH (10 mL, 2 M) and H2O (20 mL). The organic fraction was then dried (MgSO4) and removed in vacuo. The desired amide was then obtained after column chromatography on flash silica (eluent: 40-50 % EtOAc/cyclohexane or as indicated). If required, recrystallization was also performed to provide purified compounds.
N-(3-Bromophenyl)-4,5-dimethoxy-2-nitrobenzamide 13a
Obtained as a yellow-coloured semi-solid after purification by way of column chromatography (Eluent: 50 % EtOAc/cyclohex-ane); yield 73 %; 1H NMR (400 MHz, DMSO-d6) d 10.69 (s, 1H), 8.03 (d, J = 1.7 Hz, 1H), 7.73 (s, 1H), 7.62-7.56 (m, 1H), 7.35-7.31 (m, 3H), 3.94 (s, 3H), 3.93 (s, 3H); 13C NMR (101 MHz, DMSO-d6) d 164.4, 153.1, 149.1, 140.6, 138.6, 130.8, 126.9, 126.3, 121.8,121.5, 118.3, 111.1, 107.3, 56.7, 56.4; HRMS Calculated 381.0081 (M+ + H) for C15H14BrN2O5, found 381.0085.
N-(3-Chloro-4-fluorophenyl)-4,5-dimethoxy-2-nitrobenz-amide 13c
Obtained as a yellow-coloured solid after purification by way of column chromatography (Eluent: 40 % EtOAc/cyclohex-ane), followed by recrystallization from EtOAc; yield 56 %; mp 218-220 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 7.97 (dd, J = 6.8, 2.6 Hz, 1H), 7.72 (s, 1H), 7.56-7.53 (m, 1H), 7.44-7.40 (m, 1H), 7.31 (s, 1H), 3.94 (s, 3h), 3.92 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.3, 153.4 (d, J = 243.6 Hz), 153.1, 149.1, 138.6 (d, J = 3.0 Hz), 136.2,126.8,120.9,119.9 (d, J = 6.8 Hz),119.2 (d, J = 18.1 Hz), 117.2 (d, J = 21.5 Hz), 111.1,107.3,56.7,56.4; HRMS Calculated 357.0462 (M+ + H) for C15H1337ClFN2O5, found 357.0463.
N-(3,4-Dimethoxyphenyl)-4,5-dimethoxy-2-nitrobenzamide 13e
Obtained as a yellow-coloured solid after purification by way of column chromatography (Eluent: 70 % EtOAc/cyclohex-ane), followed by recrystallization from EtOAc; yield 58 %; mp 185-188 °C; 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H), 7.48 (s, 1H), 7.23 (d, J = 2.4 Hz, 1H), 6.97-6.90 (m, 2H), 6.76 (d, J = 8.7Hz, 1h), 3.94 (s, 3H), 3.92 (s, 3H), 3.84 (s, 3H), 3.80 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 166.2,153.9,149.2,148.9,146.4,138.9,132.1, 127.8,113.2,111.8,110.6,107.4,105.7,56.8,56.6,56.2, 55.9; HRMS Calculated 363.1187 (M+ + H) for C17H19N2O7, found 363.1188.
N-(3-Ethynylphenyl)-4,5-dimethoxy-2-nitrobenzamide 13f
Obtained as a yellow-coloured solid after purification by way of column chromatography (Eluent: 10 % MeOH/EtOAc); yield 68 %; mp 179-181 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 7.85 (s, 1H), 7.71 (s, 1H), 7.63 (d, J = 9.4 Hz, 1H), 7.39-7.35 (m, 1H), 7.30 (s, 1H), 7.22 (d, J = 7.6 Hz, 1H), 4.20 (s, 1H), 3.94 (s, 3H), 3.92 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.3, 153.1, 149.0, 139.2, 138.6, 129.3, 127.1, 126.9, 122.4, 122.1, 120.1, 111.1, 107.3, 83.3, 80.7, 56.7, 56.4; HRMS Calculated 327.0981 (M+ +H) for C17H15N2O5, found 327.0980.
2-[4-(4,5-Dimethoxy-2-nitrobenzoyl)-1-piperazinyl]pyrimi-dine 13 g
Obtained as white crystals (recrystalized from EtOAc); yield 94 %; mp 232-234 °C; 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 4.8 Hz, 2H), 7.72 (s, 1H), 6.77 (s, 1H), 6.55-6.51 (m, 1H), 3.98 (s, 3H), 3.97 (s, 3H), 3.99-3.95 (m, 6H), 3.25 (t, J = 5.3, 2H); 13C NMR (101 MHz, CDCl3) δ 166.8, 161.5, 157.7, 154.3, 149.2, 137.9, 126.9,110.5,109.07,107.3,56.7,56.5,46.5,43.5,43.1,41.8; HRMS Calculated 374.1460 (M+ + H) for C17H20N5O5, found 374.1459.
1-(3,4-Dichlorophenyl)-4-(4,5-dimethoxy-2-nitrobenzoyl) piperazine 13h
Obtained as a pale yellow foam (deemed pure enough after work-up); yield quantitative; 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 7.52 (d, J = 8.9 Hz, 1H), 6.91 (d, J = 2.9 Hz, 1H), 6.74 (s, 1H), 6.70 (dd, J = 2.9,8.9 Hz, 1H), 3.95 (s, 3H), 3.94 (s, 3H), 3.30-3.01 (m, 8H); 13C NMR (101 MHz, CDCl3) δ 166.5,154.3,150.2,149.2,137.8, 132.9,130.5,126.5,123.2,118.0,116.1,109.0,107.2,60.4,56.5,48.8, 48.6, 46.2, 41.5; HRMS Calculated 440.0773 (M+ +H)for C19H20Cl2N3O5, found 440.0775.
1-(4,5-Dimethoxy-2-nitrobenzoyl)-4-methylpiperazine 13i
Obtained as a pale yellow foam (deemed pure enough after work-up); yield 96 %; 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 6.73 (s, 1H), 3.95 (br s, 6H), 3.19 (t, J = 5.1 Hz, 2H), 2.56-2.48 (m, 6H), 2.30 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.3, 154.2, 149.1, 137.8, 127.0, 109.1, 107.2, 56.7, 56.5, 54.6, 54.1, 46.6, 46.0, 41.7; HRMS Calculated 310.1398 (M+ + H) for C14H20N3O5, found 310.1398.
1-Benzoyl-4-(4,5-dimethoxy-2-nitrobenzoyl)piperazine 13j
Obtained as pale yellow crystals (recrystalized from EtOAc); yield 76 %;mp185-187°C;1HNMR (400 MHz, CDCl3) δ 7.69(brs, 1H), 7.40 (br s, 5H), 6.74 (s, 1H), 4.10-4.00 (br M, 1H), 4.00-3.95 (br M, 1H), 3.96 (br s, 6H), 3.75-3.50 (br M, 4H), 3.30-3.15 (br M, 2H); 13C NMR (101 MHz, CDCl3) δ 170.6,166.7,154.4,149.3,137.8, 135.0, 130.1, 128.6, 127.0, 126.4, 108.9, 107.3, 56.7, 56.5, 46.7 (br), 42.0 (br); HRMS Calculated 400.1496 (M+ + H) for C20H22N3O6, found 400.1503.
1-(4,5-Dimethoxy-2-nitrobenzoyl)-4-(2-phenylethyl) piperazine 13k
Obtained as a pale yellow semi-solid (purified by silica gel column chromatography: 10 % MeOH-EtOAc); yield 68 %; 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 7.33-7.09 (m, 5H), 6.74 (s, 1H), 3.95 (br s, 6H), 3.95-3.90 (br M, 1H), 3.79-3.71 (br M, 1H), 3.21 (t, J = 5.1 Hz, 2H), 2.81-2.74 (br M, 2H), 2.71-2.54 (br M, 4h), 2.45-2.33 (br M, 2H); 13C NMR (101 MHz, CDCl3) δ 166.3, 154.2, 149.0,139.9,137.8,128.6,128.3,127.0,126.1,109.1,107.2,60.0,56.6, 56.5,52.7,52.1,46.6,41.8,33.5; HRMS Calculated 400.1859 (M+ + H) for C21H26N3O5, found 400.1867.
2-[4-(4,5-Dimethoxy-2-nitrobenzoyl)-1-piperazinyl] ethanol 131
Obtained as a pale yellow oil; yield 71 %, (deemed pure enough after work-up); 1H NMR (400 MHz, CD3OD) δ 7.78 (s, 1H), 6.97 (s, 1H), 3.96 (s, 3H), 3.95 (s, 3H), 3.91-3.84 (br M, 1H), 3.78-3.72 (br M, 1H), 3.69 (t, J = 5.1 Hz, 2H), 3.32-3.27 (m, 2h), 2.67 (t, J = 5.1 Hz, 2H), 2.58 (t, J = 5.8 Hz, 2H), 2.52-2.45 (m, 2h); 13C NMR (101 MHz, CD3OD) δ 169.0, 156.1, 151.0, 139.3, 127.5, 110.6, 108.5, 61.2, 59.8, 57.4, 57.0, 54.1, 53.6, 47.9, 42.8; HRMS Calculated 340.1504 (M+ + H) for C15H22N3O6, found 340.1503.
N-{2-[4-(4,5-Dimethoxy-2-nitrobenzoyl)-1-piperazinyl]ethyl}-Ν,Ν-dimethylamine 13m
Obtained as a yellow oil; yield 55 %, (deemed pure enough after work-up); 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 6.73 (s, 1H), 3.77 (s, 6H), 3.93-3.87 (br M, 1H), 3.76-3.70 (brM, 1H), 3.19 (t, J = 5.1 Hz, 2H), 2.67-2.60 (br M, 1H), 2.56-2.52 (br M, 1H), 2.51-2.47 (br M, 2H), 2.43-2.36 (br M, 4H), 2.22 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 166.3, 154.2, 149.0, 137.8, 127.0, 109.1, 107.2, 56.8, 56.7, 56.5, 56.4, 53.2, 52.6,46.6,45.8,41.8; HRMS Calculated 367.1978 (M+ + H) for C17H27N4O5, found 367.1976.
General Procedure 1: Synthesis of 2-Amino-N-phenylbenz-amides 14 by way of the Pd/C Reduction of 2-Nitro-N-phenylbenzamides 13
The 2-nitro-N-phenylbenzamides 13 (~0.5 mmol) were dissolved in EtOH (5 mL), after which 5 % Pd/C (10 % by mass) was added carefully. This was followed by the addition of ammonium formate (5. Mol equiv.), after which the reaction mixture was heated at reflux, with stirring, for 90 min. After cooling to RT, the mixture was filtered through a Celite plug and the filter cake washed with EtOH (3x5 mL). The solvent was removed to afford the anilines 14, which in general were judged by 1H NMR spectroscopy to be pure enough for biochemical evaluation; if not, flash silica gel column chromatography was employed. See individual descriptions for specific experimental details below.
2-Amino-N-(3-bromophenyl)-4,5-dimethoxybenzamide 14a
For this particular compound, the general reduction described above resulted in the formation of 14b in which the aromatic bromide had also been removed. An alternative reduction method, involved the treatment of 13a (0.060 g, 0.16 mmol) in EtOH (2 mL) with elemental iron (0.044 g, 0.80 mmol) with HCl (12 M, 0.7 mL) for 5.5 h at reflux. H2O (10 mL) was then added, after which the mixture was basified by the addition of NaOH solution (2 M). Extraction with EtOAC (3x5 mL), washing with brine (10 mL), drying with Na2SO4 and removal of solvent under reduced pressure, afforded a mixture of the desired product and starting material. Silica gel column chroma-tography (50 % EtOAc/hexanes) then resulted in 14a as a yellow semi-solid (0.010 g, 17 %), in addition to starting material 13a (20 %). The NMR spectra of 14a compared well with that published in the literature.121H NMR (400 MHz, CD3OD) d 7.98-7.94 (m, 1H), 7.63-7.57 (m, 1H), 7.28 (d, J = 1.2 Hz, 1H), 7.27-7.26 (m, 2H), 6.46 (s, 1H), 3.87 (s, 3H), 3.85 (s, 3H).
2-Amino-4,5-dimethoxy-N-phenylbenzamide 14b
For this particular compound, use of the general procedure described above resulted in removal of the bromine atom and 14b was thus obtained as a beige solid (quantitative yield). The NMR spectra of 14b compared well with that published in the literature.37 1H NMR (400 MHz, CD3OD) d 7.66 (dd, J = 7.7, 0.9 Hz, 2H), 7.44-7.41 (m, 2H), 7.33 (br s, 1H), 7.21-7.19 (m, 1H), 6.50 (br s, 1H), 3.94 (s, 3H), 3.93 (s, 3H).
2-Amino-N-(3-chloro-4-fluorophenyl)-4,5-dimethoxy-benzamide 14c
For this particular compound, the general reduction described above resulted in the formation of 14c in which the aromatic chloride had also been removed (see spectroscopic description below). An alternative reduction method, involved the treatment of 13c (0.070 g, 0.20 mmol) in EtOH (2 mL) with elemental iron (0.10 g, 2.0 mmol added in two portions at start of reaction and after 5.5 h) with HCl (12 M, 0.8 mL) for 28 h at reflux. H2O (10 mL) was then added, after which the mixture was made basic by the addition of NaOH solution (2 M). Extraction with EtOAC (3x5 mL), washing with brine (10 mL), drying with Na2SO4 and removal of solvent under reduced pressure, afforded a mixture of the desired product and starting material. Silica gel column chromatography (50 % EtOAc/hexanes) then resulted in 14c as a light yellow-coloured semi-solid (0.022 g, 35 %). The NMR spectra of 14c compared well with that published in the litera-ture.121H NMR (400 MHz, CD3OD) d 7.93-7.89 (m, 1H), 7.60-7.57 (m, 1H), 7.30 (s, 1H), 7.27-7.21 (m, 1H), 6.49 (s, 1H), 3.94 (s, 3H), 3.92 (s, 3H).
2-Amino-N-(4-fluorophenyl)-4,5-dimethoxybenzamide 14d
Obtained as pale yellow oil (purified by silica gel column chromatography: 50 % EtOAc-hexane), yield 86 %; 1H NMR (400 MHz, CD3OD) d 7.60-7.53 (m, 2H), 7.22 (s, 1H), 7.09-7.01 (m, 2H), 6.40 (s, 1H), 4.82 (s, 3H), 3.82 (s, 3H), 3.86 (s, 3H); 13C NMR (101 MHz, CD3OD) d 168.8,160.4 (d), 154.0,145.9,141.1,134.8 (d), 123.8 (d), 115.1 (d), 112.6 (d), 107.9,101.3,57.3,55.6; HRMS Calculated 291.1140 (M+ + H) for C15H16N2O3F, found 291.1140.
2-Amino-N-(3,4-dimethoxyphenyl)-4,5-dimethoxybenzamide 14e
Obtained as an off-white foam (purified by silica gel column chromatography: EtOAc), yield 43 %; 1H NMR (400 MHz, CD3OD) d 7.28 (d, J = 2.4 Hz, 1H), 7.22 (s, 1H), 7.09 (dd, J = 8.6, 2.4 Hz, 1H), 6.89 (d, J = 8.7 Hz, 1H), 6.40 (s, 1H), 4.82 (s, 3H), 3.80 (s, 3H), 3.81 (s, 3H), 3.79 (s, 3H), 3.78 (s, 3H); 13C NMR (101 MHz, CD3OD) d 169.9, 155.1, 150.3, 147.4, 147.3, 141.8, 133.6, 115.3, 114.0,113.2,108.5,108.2,101.8,57.7,56.7,56.4,56.1; HRMS Calculated 333.1445 (M+ + H) for C17H21N2O5, found 333.1446.
2-Amino-N-(3-ethynylphenyl)-4,5-dimethoxybenzamide 14f
Obtained as an off-white semi-solid (purified by silica gel column chromatography: 50 % EtOAc-hexane), yield 43 %, 1H NMR (400 MHz, CD3OD, alkyne proton not observed in spectrum) d 7.90-7.89 (m, 1H), 7.74-7.73 (m, 1H), 7.46-7.39 (m, 1H), 7.37-7.32 (m, 2H), 6.53 (s, 1H), 3.98 (s, 3H), 3.97 (s, 3H); 13C NMR (101 MHz, CD3OD) d 169.5,154.7,146.9,141.4,139.7,129.4,128.3, 125.6,123.6,122.7,113.3,108.0,101.6,84.0, 78.1,57.5, 56.1; HRMS Calculated 297.1234 (M+ + H) for C17H17N2O3, found 297.1235.
4,5-Dimethoxy-2-{[4-(2-pyrimidinyl)-1-piperazinyl]carbonyl} phenylamine 14g
Obtained as a pale yellow semi-solid, (purified by silica gel column chromatography: 5 % MeOH/EtOAc); yield 40 %; 1H NMR (400 MHz, CDCl3) d 8.31 (d, J = 4.7 Hz, 2H), 6.66 (s, 1H), 6.52 (t, J = 4.7 Hz, 1H), 6.26 (s, 1H), 4.27 (br s, 2H), 3.88-3.84 (m, 4H), 3.84 (s, 3H), 3.76 (s, 3H), 3.71-3.62 (m, 4H); 13C NMR (100 MHz, CDCl3) d 170.4,161.5,157.7,151.7, 141.3,141.1,112.2, 110.4, 110.0, 101.0, 56.7, 55.8, 45.1 (br), 43.9; HRMS Calculated 344.1717 (M+ + H) for C17H22N5O3, found 344.1718.
2-{[4-(3,4-Dichlorophenyl)-1-piperazinyl]carbonyl}-4,5-dimethoxyphenylamine 14h
Obtained as a pale beige-coloured oil, (purified by silica gel column chromatography: EtOAc), yield 69 %; 1H NMR (400 MHz, CD3OD) d 7.29 (d, J = 8.9 Hz, 1H), 7.06 (d, J = 2.9 Hz, 1H), 6.86 (dd, J = 9.0,2.9 Hz, 1H), 6.72 (s, 1h), 6.45 (s, 1H), 3.79 (s, 3H), 3.84 (s, 3H), 3.84-3.80 (m, 4H), 3.24-3.14 (m, 4H); 13C NMR (100 MHz, CD3OD, one aliphatic carbon not observed in spectrum) d 172.0, 153.5,152.1, 142.7,142.4, 133.6,131.6, 123.2,118.6, 117.1, 114.3, 111.5, 102.2, 57.6, 56.2, 49.4 (br); HRMS Calculated 410.1033 (M+ + H) for C19H22N3O3Cl2, found 410.1031.
4,5-Dimethoxy-2-[(4-methyl-1-piperazinyl)carbonyl]phenyl-amine 14i
Obtained as a pale yellow oil which slowly solidified to a semi-solid; yield quantitative; 1H NMR (400 MHz, CD3OD) d 6.69 (s, 1H), 6.45 (s, 1H), 3.80 (s, 3H), 3.73 (s, 3H), 3.64-3.59 (m, 4H), 2.53-2.40 (m, 4H), 2.31 (s, 3H); 13C NMR (100 MHz, CD3OD, one aliphatic carbon not observed in spectrum) d 180.8, 162.3,151.4, 151.2, 122.9, 120.4, 110.9, 66.4, 65.0, 64.7, 54.8; HRMS Calculated 280.1656 (M+ + H) for C14H22N3O3, found 280.1656.
2-[(4-Benzoyl-1-piperazinyl)carbonyl]-4,5-dimethoxyphenyl-amine 14j
Obtained as a light yellow oil, (purified by silica gel column chromatography:5%MeOH/EtOAc); yield 93 %; 1H NMR (400 MHz, CDCl3) d 7.38 (s, 5H), 6.60 (s, 1H), 6.23 (s, 1H), 4.48 (s, 2H), 3.79 (s, 3H), 3.74 (s, 3H), 3.77-3.44 (m, 8H); 13C NMR (100 MHz, CDCl3) d 170.6, 151.9, 141.3, 141.1, 135.1, 130.0, 128.5, 127.0, 112.1, 109.4, 101.0, 56.7, 55.7, 47.4 (br), 45.2 (br); HRMS Calculated 370.1761 (M+ + H) for C20H24N3O4, found 370.1762.
4,5-Dimethoxy-2-{[4-(2-phenylethyl)-1-piperazinyl]carbonyl} phenylamine 14k
Obtained as a yellow oil (purified by silica gel column chroma-tography: 10 % MeOH/EtOAc); yield 75 %; 1H NMR (400 MHz, CD3OD) d 7.34-7.05 (m, 5H), 6.69 (s, 1H), 6.45 (s, 1H), 3.78 (s, 3H), 3.72 (s, 3H), 3.65-3.61 (m, 4H), 2.84-2.77 (m, 2H), 2.68-2.61 (m, 2H), 2.60-2.58 (m, 4H); 13C NMR (100 MHz, CD3OD, one aliphatic carbon not observed in spectrum) d 171.8, 153.5, 142.6, 142.4, 141.0, 129.7,129.5,127.2,114.2,111.6,102.2, 61.1, 57.6, 56.2, 54.1, 45.7 (br), 33.9; HRMS Calculated 370.2125 (M+ +H)for C21H28N3O3, found 370.2127.
2-[4-(2-Amino-4,5-dimethoxybenzoyl)-1-piperazinyl]ethanol 141
Pale yellow oil; yield 96 %; 1H NMR (400 MHz, CD3OD) d 6.69 (s, 1H), 6.44 (s, 1H), 3.78 (s, 3H), 3.71 (s, 3H), 3.76-3.63 (m, 6H), 2.74-2.68 (m, 6H); 13C NMR (100 MHz, CD3OD, one aliphatic carbon not observed) d 171.8, 153.5, 142.7, 142.3, 114.2, 111.3, 111.1, 102.1, 60.8, 59.1, 57.6, 56.2, 54.2, 45.2 (br), 45.0 (br); HRMS Calculated 310.1761 (M+ + H) for C15H24N3O4, found 310.1763.
General Synthesis of [(Pyridinylmethyl)amino]benzoates 16 and 17
The methyl 2-aminobenzoates 15a or 15b (5-7 mmol) were dissolved in MeOH (96 %, ~50 mL), to which 4-pyridine carboxaldehyde (1.3 mol equiv.) was added. After the addition of AcOH (0.6 mL) the reaction mixture was stirred at RT for 3-5 days under a N2 atmosphere. NaCNBH3 (1.3 mol equiv.) was subsequently added in small increments over a period of 30 min and the reaction mixture left to stir for a further 3-4 h. The solvent was then removed under reduced pressure to afford a gummy residue. EtOAc (8 mL) was used to dissolve the residue after which the organic phase was washed sequentially with aqueous NaHCO3 (sat., 100 mL) and brine (100 mL). The organic layer was then dried (Na2SO4), filtered and reduced under vacuum. The resulting residue was purified with silica gel column chromatography (70 % EtOAc-hexane) to afford semi-solids from which the pure products 16 or 17 were obtained by recrystallization (1:1 EtOAc/hexane) - see below for individual descriptions of products obtained.
Methyl 4,5-dimethoxy-2-[(4-pyridinylmethyl)amino] benzoate 16
Obtained as a white solid; yield 65 %; mp 142-144 °C; 1H NMR (300 MHz, CDCl3): d 8.54 (d, J = 5.4 Hz, 2H), 8.24 (br t, J = 4.9 Hz, 1H), 7.40 (s, 1H), 7.28 (d, J = 5.4 Hz, 2H), 5.94 (s, 1H), 4.47 (d, J = 5.7 Hz, 2H), 3.87 (s, 3H), 3.82 (s, 3H), 3.70 (s, 3H); 13C NMR (75 MHz, CDCl3) d 168.6, 155.2, 150.1, 148.5, 147.6, 139.7, 121.9, 113.8,101.6,94.9,56.5,55.6,51.4,46.3; HRMS Calculated 303.1339 (M+ + H) for C16H19N2O4, found 303.1333.
Methyl 2-[(pyridin-4-ylmethyl)amino]benzoate 17
For this particular compound, use of the general procedure described above on substrate 15b, resulted in the product 17 being obtained as a white solid (65 %). The NMR spectra of 17 compared well with that published in the literature.91H NMR (300 MHz, CDCl3) 8.52 (d, J = 6.0 Hz, 2H), 8.30-8.26 (m, 1H), 7.92 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.24-7.39 (m, 3H), 6.61-6.57 (m, 1H), 6.44 (d, J = 8.5 Hz, 1H), 4.46^.42 (m, 2H), 3.87 (s, 3H).
General Description Involving the Synthesis of Piperazin-1-yl{2-[(pyridin-4-ylmethyl)amino]phenyl}methanones 18 and 19, from Methyl [(pyridinylmethyl)amino]benzoates 16 and 17:38
A solution of AlMe3 in toluene (1.5 mol equiv., 2 M) was added to the methyl benzoate 16 or 17 (1.0 mmol) dissolved in toluene (3.5 mL) in a round-bottomed flask. The appropriate piperazine (1.4 mol equiv.) was then added, together with an additional volume of toluene (3.5 mL). The reaction mixture was then stirred at RT for 1 h, before being stirred under heating at 110 °C for an additional 1 h. The solvent was then removed under reduced pressure. EtOAc (10 mL) was then used to dissolve the residue, after which the organic phase was washed sequentially with aqueous NaHCO3 (sat., 50 mL) and brine (50 mL). The organic layer was dried (Na2SO4), filtered and removed under reduced pressure, resulting in a dark yellow oil. This residue was purified by silica gel column chromatography (90 % EtOAc/MeOH) to obtain the desired products 18 or 19, for which the details are given below.
4,5-Dimethoxy-2-(1-piperazinylcarbonyl)-N-(4-pyridinyl methyl)aniline 18a
Obtained as a pale yellow solid; yield 61 %; mp 109-111 °C; 1H NMR (300 MHz, CDCl3, one proton not observed in spectrum) d 8.54 (d, J = 6.0 Hz, 2H), 7.30 (d, J = 6.0 Hz, 2H), 6.71 (s, 1H), 6.06 (s, 1H), 5.85 (br t, J = 6.0 Hz, 1H),4.37 (d, J = 6.0 Hz, 2H), 3.78 (s, 3H), 3.69 (s, 3H), 3.63 (br t, J = 5.9 Hz, 4H), 2.90 (br t, J = 5.9 Hz, 4H); 13C NMR (75 MHz, CDCl3) d 170.4, 151.8, 149.9, 148.9, 142.6, 140.1, 122.0, 113.3, 109.8, 97.1, 57.0, 55.6, 47.0, 46.4 (2C); HRMS Calculated 357.1921 (M+ + H) for C19H25N4O3, found 357.1920.
2-[(4-Ethyl-1-piperazinyl)carbonyl]-4,5-dimethoxy-N-(4-pyridinylmethyl)aniline 18b
Obtained as a pale yellow solid; yield 51 %; mp 55-56 °C, 1H NMR (300 MHz, CDCl3) d 8.55 (d, J = 6.0 Hz, 2H), 7.28 (d, J = 6.0 Hz, 2H), 6.71 (s, 1H), 6.05 (s, 1h), 5.87 (br t, J = 6.1 Hz, 1H), 4.37 (d, J = 6.1 Hz, 2H), 3.78 (s, 3H), 3.72-3.68 (m, 7H), 2.50-2.43 (m, 6H), 1.09 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) d 170.4, 152.0,150.1,148.9,142.9,140.1,122.0,113.5,109.7,97.2,57.1,55.7, 53.1, 52.3,47.1,45.5 (br), 12.0; HRMS Calculated 385.2234 (M+ + H) for C21H29N4O3, found 385.2235.
4,5-Dimethoxy-N-(4-pyridinylmethyl)-2-{[4-(2-pyridinyl)-1-piperazinyl]carbonyl}aniline 18c
Obtained as an orange oil; yield 83 %; 1H NMR (300 MHz, CDCl3): d 8.68 (d, J = 4.5,2H), 8.22 (br d, J = 5.2 Hz, 1H), 7.55-7.50 (m, 1H), 7.30-7.28 (d, J = 6.0 Hz, 2H), 6.78-6.76 (m, 1h), 6.71-6.67 (m, 2H), 6.07 (s, 1H), 6.03-5.99 (m, 1H), 4.44 (d, J = 7.4 Hz, 2H), 3.79-3.77 (m, 7H), 3.70 (s, 3H), 3.64-3.60 (m, 4H); 13C NMR (75 MHz, CDCl3): d 170.8, 159.1, 152.2, 150.1, 148.7, 148.0,143.2, 140.1, 137.7, 122.0, 114.0, 113.5, 109.3, 107.3, 97.3, 57.0, 55.7, 47.1, 45.6, 45.2; HRMS Calculated 434.2187 (M+ + H) for C24H28N5O3, found 434.2184.
4,5-Dimethoxy-N-(4-pyridinylmethyl)-2-{[4-(4-pyridinyl)-1-pi perazinyl]carbonyl}aniline 18d
Obtained as a dark yellow oil; yield 22 %; 1H NMR (300 MHz, CDCl3): d 8.54 (d, J = 4.8 Hz, 2H), 8.31 (d, J = 5.3 Hz, 2H), 7.31 (d, J = 5.2 Hz, 2H), 6.74 (s, 1H), 6.69 (br s, 2H), 6.08 (br s, 2h), 4.39 (br s, 2H), 3.80-3.79 (br M, 7H), 3.71 (s, 3H), 3.42 (br s, 4H); 13C NMR (75 MHz, CDCl3): d 170.9, 154.7, 152.4, 150.1, 150.0, 148.7, 143.4, 140.1, 122.0, 113.6, 108.7, 108.6, 97.2, 57.1, 55.7, 47.0, 46.2, 44.7; HRMS Calculated 434.2187 (M+ + H) for C24H28N5O3, found 434.2183.
4,5-Dimethoxy-N-(4-pyridinylmethyl)-2-{[4-(2-pyridinyl)-1-piperazinyl]carbonyl}aniline 18e
Obtained as a dark yellow oil; yield 83 %; 1H NMR (300 MHz, CDCl3): d 8.54 (d, J = 4.5 Hz, 2H), 8.34 (d, J = 4.8 Hz, 2H), 7.30 (d, J = 6.0 Hz, 2H), 6.75 (s, 1H), 6.56-6.53 (m, 1H), 6.07 (s, 1H), 6.03-5.99 (m, 1H), 4.39 (d, J = 5.3 Hz, 2H), 3.92-3.89 (br M, 4h), 3.79 (s, 3H), 3.76-3.72 (m, 4H), 3.70 (s, 3H); 13C NMR (75 MHz, CDCl3): d 170.8,161.6,157.8,152.1,150.0,148.8,143.1,140.2,122.0, 113.5, 110.5, 109.3, 97.2, 57.0, 56.2, 47.1, 45.3, 44.0; HRMS Calculated 435.2139 (M+ + H) for C23H27N6O3, found 435.2135.
4,5-Dimethoxy-2-{[4-(2-phenylethyl)-1-piperazinyl] carbonyl}-N-(4-pyridinylmethyl)aniline 18f
Obtained as a yellow oil; yield 45 %; 1H NMR (400 MHz, CDCl3) d 8.53 (d, J = 5.8 Hz, 2H), 7.28 (d, J = 4.9 Hz, 4H), 7.22-7.18 (m, 4H), 6.70 (s, 1H), 6.04 (s, 1H), 4.36 (s, 2H), 3.77 (d, J = 3.2 Hz, 4H), 3.68 (d, J = 4.0 Hz, 6H), 2.83-2.79 (m, 2H), 2.68-2.63 (m, 2H), 2.57-2.54 (m, 4H); 13C NMR (101 MHz, CDCl3) d 170.8, 152.3, 150.9, 150.4, 149.32, 143.2, 140.4, 140.2, 129.1, 128.8, 126.6, 122.4, 113.8, 109.9, 97.5, 60.6, 57.4, 56.1, 53.8, 47.5, 33.8; HRMS Calculated 461.2547 (M+ + H) for C27H33N4O3, found 461.2543.
2-(1-Piperazinylcarbonyl)-N-(4-pyridinylmethyl)aniline 19a
Obtained as a pale yellow solid; yield 54 %; mp 84-86 °C, 1H NMR (300 MHz, CDCl3) d 8.53 (d, J = 6.0 Hz, 2H), 7.26 (d, J = 6.0 Hz, 2H), 7.18-7.10 (m, 2H), 6.72-6.67 (m, 1H), 6.47 (d, J= 9.0 Hz, 1H), 5.76 (br t, J = 6.1,1H), 4.39 (d, J = 6.1 Hz, 2H), 3.64 (br s, 4H), 2.91-2.87 (m, 4H); 13C NMR (75 MHz, CDCl3, one carbon not observed in aliphatic region of spectrum) d 170.2, 150.0, 148.6, 146.4,130.9, 128.0,121.9, 119.3,116.5, 111.9,46.5,46.4 (br); HRMS Calculated 297.1710 (M+ + H) for C17H21N4O, found 297.1709.
2-[(4-Ethyl-1-piperazinyl)carbonyl]-N-(4-pyridinylmethyl) aniline 19b
Obtained as a yellow solid; yield 45 %; mp 142-144 °C; 1H NMR (300 MHz, CDCl3) d 8.54-8.52 (m, 2H), 7.27 (d, J = 5.9 Hz, 2H,), 7.18-7.10 (m, 2H), 6.68 (t, J = 5.9 Hz, 1H), 6.47 (d, J = 9.0 Hz, 1h), 5.78 (br t, J = 6.0 Hz, 1h), 4.38 (d, J = 6.0 Hz, 2H), 3.69 (br s, 4h), 2.50-2.42 (m, 6H), 1.11 (t, J = 6.0 Hz, 3H); 13C NMR (75 MHz, CDCl3, one carbon not observed in aliphatic region) d 170.0, 150.0,148.6,146.4,131.0,128.1,122.0,119.2,116.5,111.9,53.0,52.3, 46.4, 11.9; HRMS Calculated 325.2023 (M+ + H) for C19H25N4O, found 325.2021.
N-(4-Pyridinylmethyl)-2-{[4-(2-pyridinyl)-1-piperazinyl] carbonyl}aniline 19c
Obtained as a yellow solid; yield 30 %; mp 62-64 °C; 1H NMR (300 MHz, CDCl3) d 8.52 (d, J = 5.3 Hz, 2H), 8.20 (d, J = 3.7 Hz, 1H), 7.50-7.48 (m, 1H), 7.25 (d, J = 4.6 Hz, 2H), 7.20-7.14 (m, 2H), 6.73-6.61 (m, 3H), 6.49 (d, J = 8.2 Hz, 1H), 5.88-5.85 (m, 1H), 4.37 (d, J = 5.8 Hz, 2H), 3.79 (br s, 4H), 3.61 (br s, 4H); 13C NMR (75 MHz, CDCl3, one aliphatic carbon not observed in spectrum) d 170.5, 159.1, 150.0, 148.5, 148.1, 146.6, 137.7, 131.2, 128.2,121.9, 118.9, 116.6, 114.1, 112.1, 107.3, 46.4, 45.7; HRMS Calculated 374.1981 (M+ + H) for C22H24N5O, found 374.1975.
N-(4-Pyridinylmethyl)-2-{[4-(4-pyridinyl)-1-piperazinyl] carbonyl}aniline 19d
Obtained as a pale yellow solid; yield 34 %; mp 78-80 °C; 1H NMR (300 MHz, CDCl3) d 8.54 (br s, 2H), 8.33 (br s, 2H), 7.30-7.14 (br M, 4H), 6.70 (br s, 3H), 6.52 (d, J = 5.9 Hz, 1H), 5.96 (br s, 1H), 4.40 (br s, 2H), 3.83 (br s, 4H), 3.43 (br s, 4H); 13C NMR (75 MHz, CDCl3, one aliphatic carbon not observed in spectrum) d 170.6, 154.6, 150.4, 150.0, 148.4, 146.8, 131.5, 128.2, 121.9,118.2, 116.5, 112.2, 108.7, 46.4, 46.3; HRMS Calculated 374.1981 (M+ +H) for C22H24N5O, found 374.1983.
Activity Based Assay for IC50 Determination
IC50 determinations for EGFR wt, L858R and T790M/L858R (purchased from Invitrogen: PV3872, PV4128, PV4879) were measured with the HTRF KinEASE-TKassay from Cisbio according to the manufacturer's instructions. After 2 hours of pre-incubation with the tested inhibitor and 50 nM of an artificial biotinylated substrate peptide (TK-substrate), EGFR was allowed to phosphorylate the latter by adding an ATP concentration corresponding to its KM (30 µM for EGFR wt, 60 for EGFR L858R and 30 µM for EGFR T790M/L858R, previously determined using the same assay and EGFR constructs). After completion of the reaction, an antiphosphotyrosine antibody labelled with Europium cryptate and streptavidin labelled with the fluorophore XL665 were added. The FRET between Europium cryptate and XL665 was measured to quantify the phos-phorylation of the substrate peptide (Tecan Safire 2 plate reader, excitation at 317 nm, readout at 620 nm -Eu-labelled antibody-and 665 nm -XL665 labelled streptavidin- after 60 ,us lag time). The quotient of both intensities for reactions made with eight different inhibitor concentrations (including no inhibitor) were plotted against inhibitor concentrations and fit to a Hill 4-parameter equation to determine IC50 values (IDBS XLfit). Each reaction was performed in duplicate, and at least three independent determinations of each IC50 were made.
According to the instructions given for EGFR testing, Akt1 wt (120 pM, Millipore, Lot # D8MN034U-L), Akt2 wt (501 pM, Invitrogen, Lot # PV3184_28770N) and ΔPH-Aktl (Millipore, Lot # 1600485-E), respectively were pre-incubated with the respective inhibitors (eight different concentrations) in a dark wet chamber for1hatRT. The phosphorylation reaction was started by adding both ATP (50 µM for Akt1,65 for both Akt 2 and ΔPH-Akt1) and STK-substrate 3 (250 nM for Akt1,300 nM for both Akt2 and ΔPH-Akt1). After incubation for 45 min (Akt 1), 20 min (Akt2) or 17 min (ΔPH-Akt1), respectively the reaction was stopped and further incubated for1hatRT. Each well was excited at 317 nm and emission was measured at 620 nm and 665 nm with a 60 ,s delay using a Tecan infinite M1000 plate reader.
References and Notes
1 C. Congiu, M.T. Cocco, V Lilliu and V Onnis, J. Med. Chem., 2005,48, 8245-8252. [ Links ]
2 V Onnis, M.T. Cocco, V. Lilliu and C. Congiu, Bioorg. Med. Chem, 2008, 16, 2367-2378. [ Links ]
3 M.T. Cocco, C. Congiu, V. Lilliu and V Onnis, Bioorg. Med. Chem. Lett., 2004,14, 5787-5791. [ Links ]
4 M.T. Cocco, C. Congiu, V. Onnis, M. Morelli, V. Felipo and O. Cauli, Bioorg. Med. Chem., 2004,12, 4169^177. [ Links ]
5 S.D. Barrett, A.J. Bridges, D.T. Dudley, A.R. Saltiel, J.H. Fergus, C.M. Flamme, A.M. Delaney, M. Kaufman, S. LePage, W.R. Leopold, S.A. Przybranowski, J. Sebolt-Leopold, K. Van Becelaere, A.M. Doherty, R.M. Kennedy, D. Marston, W.A. Howard, Jr., Y. Smith, J.S. Warmus and H. Tecle, Bioorg. Med. Chem. Lett., 2008,18, 6501-6504. [ Links ]
6 P. Wickens, H. Kluender, J. Dixon, C. Brennan, F. Achebe, A. Bacchiocchi, D. Bankston, D. Bierer, M. Brands, D. Braun, M.S. Brown, C.-Y. Chuang, J. Dumas, I. Enyedy, G. Hofilena, Z. Hong, T. Housley, B. Jones, U. Khire, C. Kreiman, E. Kumarasinghe, T. Lowinger, R. Ott-Morgan, L. Perkins, B. Phillips, R. Schoenleber, W.J. Scott, R. Sheeler, A. Redman, X. Sun, I. Taylor, L. Wang, S. Wilhelm, X. Zhang, M. Zhang, E. Sullivan, C. Carter, M. Miglarese and J. Levy, Bioorg. Med. Chem. Lett, 2007,17, 4378^381. [ Links ]
7 A. Papakyriakou, M.E. Katsarou, M. Belimezi, M. Karpusas and D. Vourloumis, ChemMedChem, 2010, 5, 118-129. [ Links ]
8 M. Rogosnitzky, R. Danks and E. Kardash, Anticancer Res., 2012, 32, 2471-2478. [ Links ]
9 P.W. Manley, P. Furet, G. Bold, J. Bruggen, J. Mestan, T. Meyer, C.R. Schnell and J. Wood, J. Med. Chem., 2002, 45, 5687-5693. [ Links ]
10 F. Musumeci, M. Radi, C. Brullo and S. Schenone, J. Med. Chem, 2012, 55, 10797-10822. [ Links ]
11 T. Asano, T. Yoshikawa, H. Nakamura, Y. Uehara and Y. Yamamoto, Bioorg. Med. Chem. Lett., 2004, 14, 2299-2302. [ Links ]
12 T. Asano, T. Yoshikawa, T. Usui, H. Yamamoto, Y. Yamamoto, Y. Uehara and H. Nakamura, Bioorg. Med. Chem, 2004,12,3529-3542. [ Links ]
13 S. Kamath and J.K. Buolamwini, Med. Res. Rev, 2006, 26, 569-594. [ Links ]
14 H. Nakamura, Y. Sasaki, M. Uno, T. Yoshikawa, T. Asano, H.S. Ban, H. Fukazawa, M. Shibuya and Y. Uehara, Bioorg. Med. Chem. Lett., 2006, 16, 5127-5131. [ Links ]
15 P. Furet, G. Bold, F. Hofmann, P. Manley, T. Meyer and K.H. Altmann, Bioorg. Med. Chem. Lett., 2003, 13, 2967-2971. [ Links ]
16 P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer, Bioorg. Med. Chem. Lett, 2008,18, 897-900. [ Links ]
17 B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601-2611. [ Links ]
18 A. Kamal, E.V. Bharathi, M.J. Ramaiah, J.S. Reddy, D. Dastagiri, A. Viswanath, F. Sultana, S. Pushpavalli, M. Pal-Bhadra, A. Juvekar, S. Sen and S. Zingde, Bioorg. Med. Chem. Lett, 2010, 20, 3310-3313. [ Links ]
19 A. Kamal, J.R. Tamboli, M.J. Ramaiah, S.F. Adil, G.K. Rao, A. Viswanath, A. Mallareddy, S.N.C.V.L. Pushpavalli and M. Pal-Bhadra, ChemMedChem, 2012, 7, 1453-1464. [ Links ]
20 Kamal, A. and Prasad, B.R., Patent, International patent number: WO 2008/114275 A2, 2008, Novel anthranilic acid derivatives as potential anticancer agents and a process for the preparation thereof. [ Links ]
21 M. Getlik, C. Grütter, J.R. Simard, H.D. Nguyen, A. Robubi, B. Aust, W.A.L. van Otterlo and D. Rauh, Eur. J. Med. Chem, 2012, 48, 1-15. [ Links ]
22 V.G. Pawar, M.L. Sos, H.B. Rode, M. Rabiller, S. Heynck, W.A.L. van Otterlo, R.K. Thomas and D. Rauh, J. Med. Chem., 2010,53,2892-2901. [ Links ]
23 R. Schneider, A. Gohla, J.R. Simard, D.B. Yadav, Z. Fang, W.A.L. van Otterlo and D. Rauh, J. Am. Chem. Soc., 2013,135, 8400-8408. [ Links ]
24 M. Getlik, J.R. Simard, M. Termathe, C. Grütter, M. Rabiller, W.A.L. van Otterlo and D. Rauh, PLOS One, 2012, 7, e39713. [ Links ]
25 S. Klüter, J.R. Simard, H.B. Rode, C. Grütter, V. Pawar, H.C.A. Raaijmakers, T.A. Barf, M. Rabiller, W.A.L. van Otterlo and D. Rauh, ChemBioChem, 2010,11, 2557-2566. [ Links ]
26 R. Ul Islam, J. Hean, W.A.L. van Otterlo, C.B. de Koning and P. Arbuthnot, Bioorg. Med. Chem. Lett., 2009,19, 100-103. [ Links ]
27 R. Ul Islam, M.M. Johnson, R. Mohammad, J. Hean, P. Arbuthnot, C.B. de KoningandW.A.L. van Otterlo, S. Afr. J. Chem.,2010,63,88-94. [ Links ]
28 J.-C. Soria, T.S. Mok, F. Cappuzzo and P.A. Jaenne, Cancer Treat. Rev., 2012, 38, 416-430. [ Links ]
29 P.C. Ma, Cleve. Clin. J. Med., 2012, 79, eS56-eS60. [ Links ]
30 G.J. Riely, J. Thorac. Oncol., 2008, 3, S146-S149. [ Links ]
31 P. Warnault, A. Yasri, M. Coisy-Quivy, G. Cheve, C. Bories, B. Fauvel and R. Benhida, Curr. Med. Chem, 2013, 20, 2043-2067. [ Links ]
32 A. Richters, J. Ketzer, M. Getlik, C. Grütter, R. Schneider, J.M. Heuckmann, S. Heynck, M.L. Sos, A. Gupta, A. Unger, C. Schultz-Fademrecht, R.K. Thomas, S. Bauer and D. Rauh, J. Med. Chem., 2013, 56, 5757-5772. [ Links ]
33 See http://www.kinase-screen.mrc.ac.uk/ for information concerning this commercial screening platform. [ Links ]
34 Both compounds were resent to Dundee for retesting and this time activities measured were significantly less [IC50s: 14a (40 % @ 10 µΜ) and 14h (69 % @ 10 µ [ Links ]Μ].
35 See http://www.reactionbiology.com for information concerning this commercial screening platform. [ Links ]
36 Extrapolation of the Reaction Biology dose-response data for Aurora B would suggest an IC50 of 459 µΜ for 14a, although the Dundee screen would imply an IC50 of about 10 µΜ. This discrepancy might be explained by the ATP-Km of the Aurora B used by Reaction Biology, which is about one order of magnitude higher than that of the Dundee Aurora B. [ Links ]
37 T.A. Stroganova, V.K. Vasilin, E.A. Zelenskaya, VM. Red'kin and G.D. Krapivin, Synthesis, 2008, 3088-3098. [ Links ]
38 W.A.L. van Otterlo, J.P. Michael and C.B. de Koning, Synth. Commun., 2007, 37, 3611-3621 [ Links ]
Received 30 December 2013
Revised 15 February 2014
Accepted 13 March 2014
* Authors for correspondence. E-mail: daniel.rauh@tu-dortmund.de / wvo@sun.ac.za
Appendix
Online supplement to: S. Chakravorty, H.F. Klein, L.E. Hodson, M. Rabillier, Z. Fang, A. Richters, S.C. Pelly, D. Rauh and W.A.L. van Otterlo, S. Afr. J. Chem., 2014, 67, 71-79.
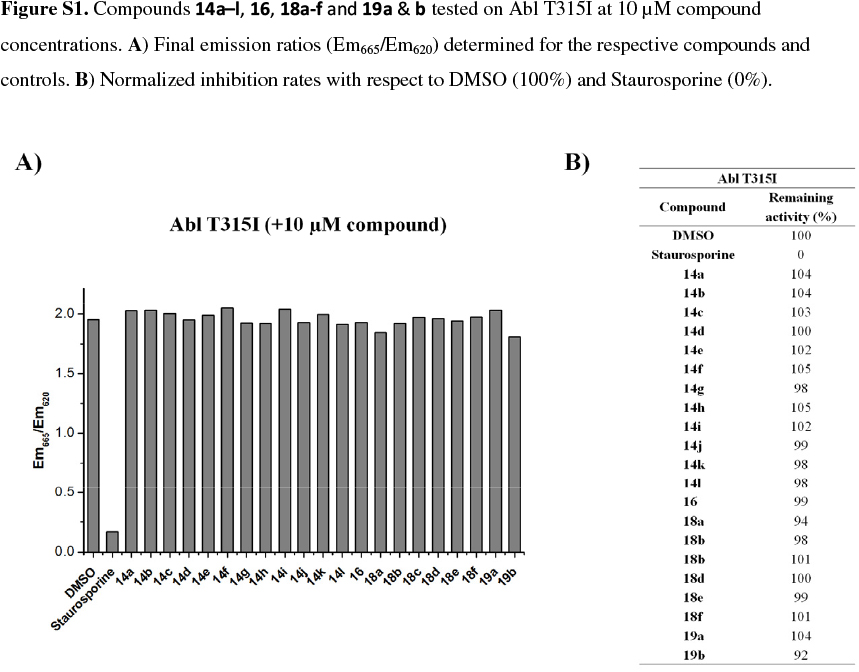
Abl T315I (0.3 ng/well), purchased from Invitrogen (Lot#39639B, PV3866), was measured with the KinEASE-TK assay from Cisbio according to the manufacturer's instructions. A biotinylated poly-Glu-Tyr substrate peptide was phosphorylated and after completion of the reaction, an anti-phosphotyrosine antibody labeled with europium cryptate and streptavidin labeled with the fluorophore XL665 were added. FRET between europium cryptate and XL665 was measured to quantify the phosphorylation of the substrate peptide. ATP concentrations were set at the KM value (6 µM) and 250 nM TK-substrate were used. Kinase and inhibitor were preincubated for 30 min before the reaction was started by addition of ATP and substrate peptide. A Tecan infinite M1000 plate reader was used to measure the fluorescence of the samples at 620 nm (Eu-labeled antibody) and 665 nm (XL665 labeled streptavidin) 60 us after excitation at 317 nm. The experiment was performed in duplicates using plain DMSO as negative control (100% remaining activity) and 10 uM Staurosporine as positive control (0% remaining activity).
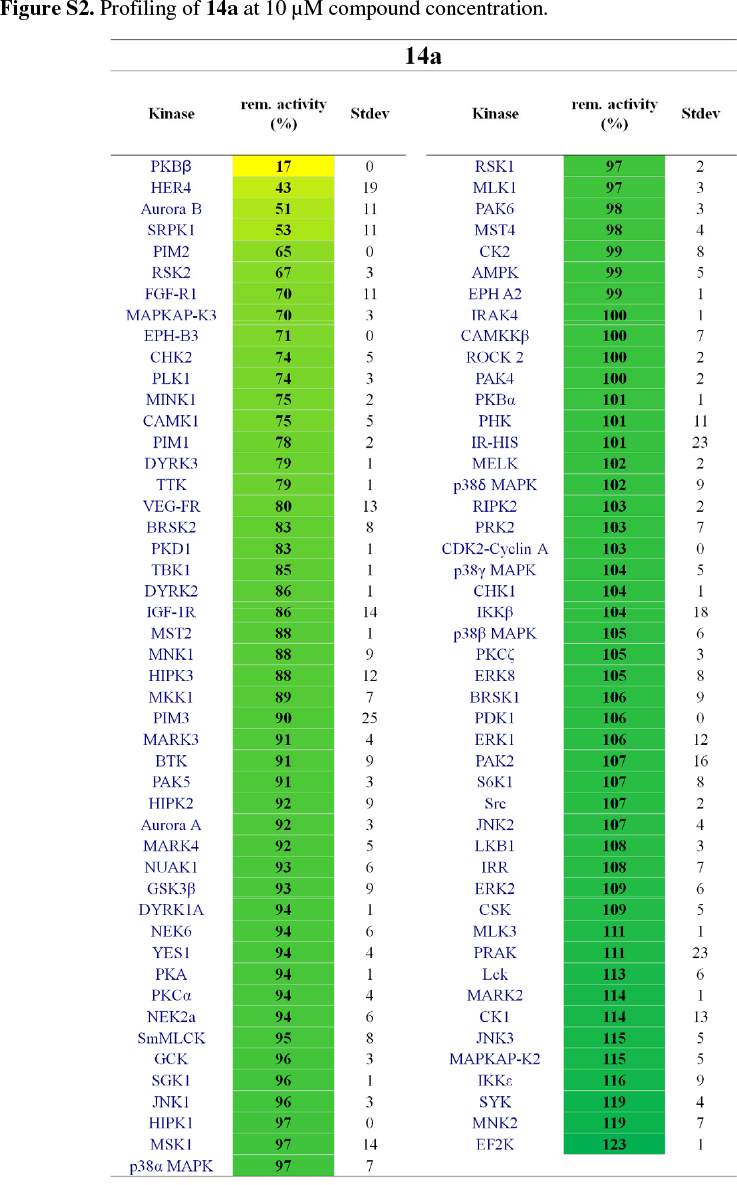
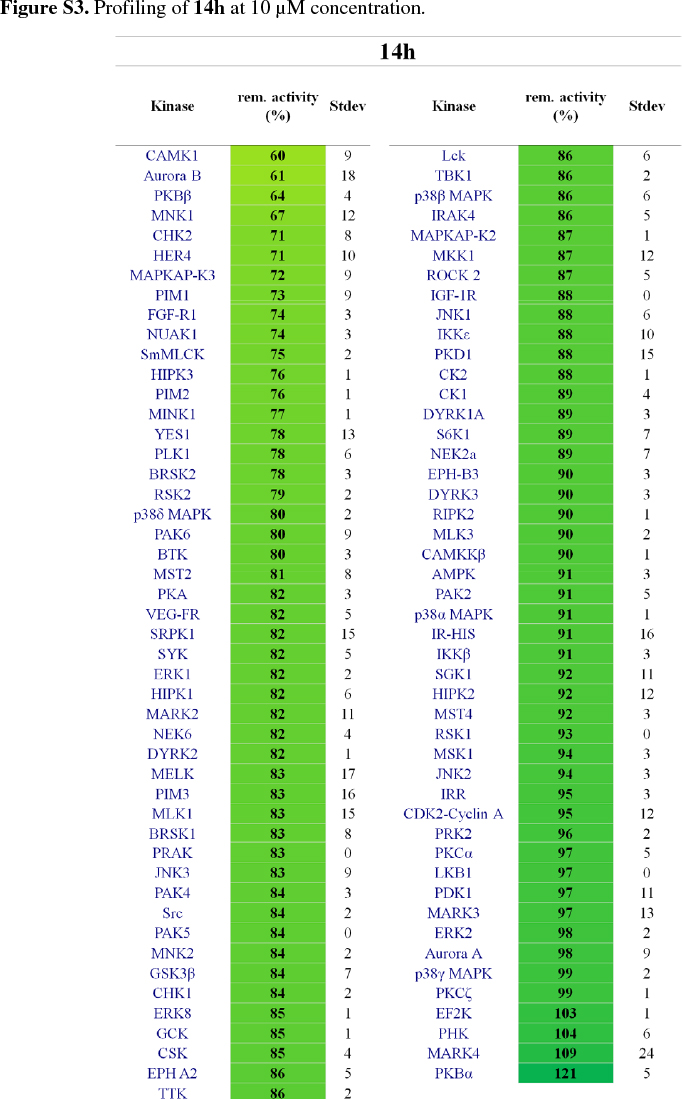
14a and 14h were tested on 95 kinases at 10 µM concentrations using a radiometric (33P-y-ATP) filter-binding assay conducted at the MRC Protein Phosphorylation Unit of the University of Dundee (http://www.kinase-screen.mrc.ac.uk/). ATP concentrations were chosen individually for each kinase to be at their respective ATP-Km. The remaining activity of each kinase was normalized and is represented in %.